«ты особенный малыш»: что делать, если у ребенка миодистрофия дюшенна
Содержание:
- Психотерапия
- Стандартные методы лечения
- Клиническая картина
- Как диагностируют заболевания?
- Диагностика мышечной дистрофии
- Физиотерапия
- Диагностика миопатии Дюшена в Германии
- Ваня выздоровел
- Диагноз
- Причина
- Другие формы заболевания
- Лечение миопатии и амиотрофии – наша работа. Клиника “Эхинацея”
- Патогенез
- Мышечная дистрофия
- Этиология возникновения паралича Дюшена-Эрба
- Благотворительные фонды помогают приехать на лечение в Израиль
- Патологическая анатомия
- Как диагностировать заболевание?
- Лекарства при миодистрофии Дюшенна
Психотерапия
Начало клинических проявлений и последующие ухудшения, как правило, провоцируются психическим стрессом. Более того, тяжелое заболевание мышц, само по себе, серьезный психический стресс. На фоне болезни часто развиваются вторичные депрессии с апатией и нежеланием заниматься своим здоровьем. А это ведет к прогрессированию болезни и возможной гибели пациента.
Если это нужно, мы проводим нашим пациентам курс психотерапевтического лечения. Наши пациенты становятся более устойчивыми к психическим раздражителям, становятся активнее, позитивно относятся к лечению, и это незамедлительно сказывается на работе мышц.
Кроме того, мы обучаем пациентов специальным психотехникам для работы с собственными мышцами.
При наличии признаков депрессии возможно назначение современных антидепрессантов (селективные ингибиторы обратного захвата серотонина, такие как Ципралекс, Флуоксетин и др.). Эти антидепрессанты не снижают, а повышают активность пациента и настроение, не вызывают мышечной слабости.
Стандартные методы лечения
Болезнь была описана еще в XIX веке неврологом Гийомом Дюшенном, в честь которого она и названа. И с того времени врачи ищут эффективные способы терапии. Первым методом, который используется до сих пор, стало применение ортопедических устройств.
Тренировка мышц помогает немного замедлить процесс их разрушения. Для пациентов применяется как активная (пока они сами в состоянии заниматься), так и пассивная тренировка. Врачи отмечают, что регулярные упражнения способны продлить период, когда пациент передвигается без инвалидной коляски.
По статистке, практически половина больных миодистрофией переживают операцию на позвоночнике. При слабых мышцах скелет искривляется, что существенно ухудшает течение болезни. Поэтому на помощь таким больным приходят различные ортопедические приспособления — фиксация костей и суставов позволяет замедлить их деформацию.
Современные разработки в этом направлении терапии давно вышли за пределы простых поддерживающих средств. Так, разработанная нидерландским Университетом Твенте роботизированная рука A-Gear способна заменить человеку атрофированную конечность. Искусственная рука улавливает электрические сигналы от мышц или минимальное мышечное напряжение. Люди с миодистрофией Дюшенна могут управлять механической конечностью, движения при этом получаются очень точными и естественными. «К исследованию мы привлекли много участников, которые уже на протяжении 3-5 лет утратили способность двигать руками, — говорит ученый Джоан Лобо-Прат (Joan Lobo-Prat). — A-Gear помогла им частично вернуть функциональность конечностей».
Клиническая картина
Как диагностируют заболевания?
Поскольку мышечные дистрофии относятся к редким патологиям, их диагностика часто бывает затруднена. К тому же эти заболевания зачастую развиваются постепенно, на протяжении многих лет или даже десятилетий, и больные подолгу не обращаются за медицинской помощью. В то же время, если своевременно начать правильное лечение, прогрессирование миодистрофий можно замедлить4.
Чтобы вовремя выявить болезнь, важно при появлении мышечной слабости обратиться к врачу, который назначает первичную диагностику и при необходимости направляет к генетику. Диагноз устанавливают на основании результатов комплексного обследования, в состав которого могут входить4:
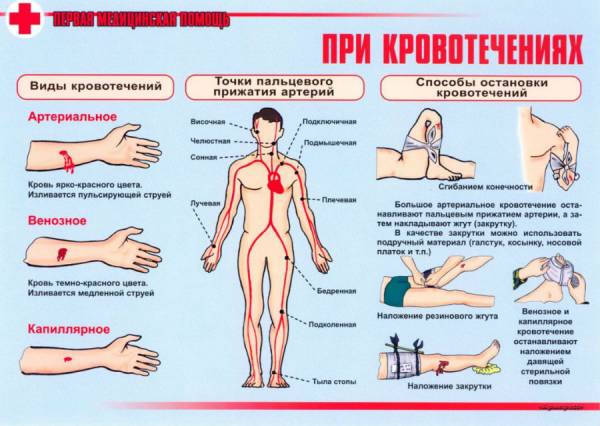
- электромиография, оценивающая электрическую активность мышц;
- биопсия мышц, позволяет отличить миодистрофию от других мышечных заболеваний;
- анализ ДНК, выявляющий мутации и другие исследования.
Диагностика мышечной дистрофии
- Аппаратные исследования: МРТ мышечной ткани применяется для определения степени поражений, ЭНМГ (электронейромиография) позволяет оценить функциональные характеристики периферической НС и мышц.
- Лабораторная диагностика: проводится биохимический АК на КФК (уровень креатинфосфокиназы) – увеличенная до 50 раз активность КФК при миодистрофических поражениях свидетельствует о прогрессировании заболевания.
Но наиболее показательными и информативными являются генетические молекулярные исследования (панель «Нервно-мышечные заболевания», микроматричный анализ – ХМА), которые точно определяют нозологическую форму мышечной дистрофии.
Кроме того, около трети диагностируемых случаев миодистрофии Дюшенна имеют спорадический характер. Возникновение 6-7% спонтанных мутаций обусловлено органным гонадным мозаицизмом – наличием у матери генетически здоровых и мутантных первичных половых клеток.
Поэтому пренатальная молекулярная диагностика позволяет идентифицировать, какой именно структурный вариант гена (аллель) получил ребенок. Даже если установлен мужской пол эмбриона, вероятность передачи от матери мутантной популяции гамет не оценивается в 100%, как и генетический риск наследования идентичной мутации братьями (сестрами) больного со спорадической миодистрофией.
Физиотерапия
В результате длительного течения болезни на месте погибших мышечных волокон образуются включения жировой и фиброзной (рубцовой) ткани, которые препятствуют работе мышц и даже могут вызывать мышечную боль. Для рассасывания этих включений и применяется физиотерапия.
Кроме того, при болезнях мышц могут формироваться контрактуры – снижение объема движений в суставах и мышцах. Здесь физиотерапия – так же один из основных методов лечения.
В нашей клинике применяются, в основном, 2 метода физиотерапии: обертывания и электрофорез с ферментными (протеолитическими) препаратами. Протеолитики оказывают мощное рассасывающее действие на фиброзную и поврежденную ткань, не затрагивая здоровых мышечных волокон. Для обертываний используется состав на основе натуральной желчи, а для электрофореза – на основе папаина.
Электростимуляция мышц применяется лишь в некоторых случаях, когда отдельные мышцы настолько слабы, что невозможно выполнение даже элементарных движений. Электростимулятор заставляет мышцы сокращаться независимо от наличия/отсутствия произвольного усилия пациента.
Мы выполняем физиолечение в клинике и обучаем пациентов самостоятельному выполнению физиопроцедур в домашних условиях. Стоимость простого аппарата для электрофореза в магазинах медтехники 3700-4000 рублей, а электромиостимулятора от 3000 рублей.
Диагностика миопатии Дюшена в Германии

осмотр ребенка у врача
После поступления пациентов в немецкие клиники специалисты проводят опрос их жалоб, анамнеза заболевания и жизни, а также общий осмотр
Для лечащего врача очень важно выявить генетическую предрасположенность у пациента к миопатии Дюшена. Поэтому очень часто ребенка обследуют вместе с мамой (которая является носителем патологического гена)
В обязательном порядке больному ребенку назначаются консультации кардиолога, ортопеда и невролога.
Для подтверждения основного диагноза используются следующие лабораторные и инструментальные методы исследований:
- Развернутый анализ крови;
- Общий анализ мочи;
- Биохимическое исследование крови с обязательным определением концентрации общего билирубина и его фракций, мочевины, мочевой кислоты, креатинина, общего белка. Особым значением в диагностике миопатии Дюшена является повышение концентрации креатинфосфокиназы (специфический фермент, который повышается при повреждении мышечной ткани). КФК повышается не только у больного ребенка, но и его у мамы;
- Рентгенологическое исследование органов грудной полости;
- УЗИ органов брюшной полости и забрюшинного пространства;
- ЭКГ – отмечается гипертрофия левого желудочка и различные нарушения ритма сердца;
- Определение концентрации дистрофина в мышечной ткани;
- Биопсия мышц с последующим гистологическим исследованием биоптата. При этом специалист должен проводить исследование только на наименее поврежденных мышцах, в противном случае исследование будет неинформативным;
- Электромиография;
- Консультация генетика с кариотипированием.

УЗИ брюшной полости
Ваня выздоровел
В Украине Ване Волохатюку в течение 2 лет не могли поставить диагноз. В МЦ Хадасса у него обнаружили редкое и очень опасное заболевание – синдром IPEX. Ребенка спасли, проведя успешную трансплантацию костного мозга (ТКМ).
Фото слева: так Ваня выглядел до того, как профессор Полина Степенски успешно провела ему ТКМ в медицинском центре Хадасса.
Фото справа: Ваня после ТКМ с сотрудницей нашего медицинского департамента Эстер Винокуров.
Для справки:
- По разным оценкам в России к 2017 году было зарегистрировано от 1.5 до 5 миллионов больных с редкими заболеваниями.
- В 2017 году в Украине на лечение орфанных заболеваний был выделен 1 млрд. гривен, и эта сумма почти сразу же была признана недостаточной.
- В Беларуси зарегистрированы тысячи случаев орфанных заболеваний, по оценкам местной прессы ими болеют от 6% до 8% населения.
Диагноз
Причина
МДД наследуется рецессивно с Х-хромосомой.
МДД вызывается мутацией гена дистрофина в локусе Xp21, расположенном на коротком плече Х-хромосомы. Дистрофин отвечает за соединение актинового цитоскелета каждого мышечного волокна с подлежащей базальной пластиной ( внеклеточным матриксом ) через белковый комплекс, содержащий множество субъединиц. Отсутствие дистрофина позволяет избытку кальция проникать через сарколемму (клеточную мембрану). Изменения в кальции и сигнальных путях заставляют воду попадать в митохондрии, которые затем лопаются.
При дистрофии скелетных мышц дисфункция митохондрий приводит к усилению индуцированных стрессом сигналов цитозольного кальция и усилению индуцированной стрессом продукции активных форм кислорода . В сложном каскадном процессе, который включает несколько путей и не совсем понятен, повышенный окислительный стресс внутри клетки повреждает сарколемму и в конечном итоге приводит к гибели клетки. Мышечные волокна подвергаются некрозу и в конечном итоге заменяются жировой и соединительной тканью .
МДД наследуется по Х-сцепленному рецессивному типу . Самки обычно являются носителями генетического признака, в то время как самцы страдают. Женщина-носитель не будет знать, что она является носителем мутации, пока у нее не появится больной сын. Сын матери-носителя имеет 50% шанс унаследовать дефектный ген от своей матери. Дочь матери-носителя имеет 50% шанс быть носителем и 50% шанс иметь две нормальные копии гена. Во всех случаях здоровый отец либо передает нормальный Y своему сыну, либо нормальный X своей дочери. Женщины-носители Х-сцепленного рецессивного состояния, такого как МДД, могут проявлять симптомы в зависимости от их модели X-инактивации . МДД встречается у каждого 3600 младенцев мужского пола. Мутации в гене дистрофина могут быть наследственными или возникать спонтанно во время передачи по зародышевой линии.
МДД крайне редко встречается у женщин (примерно у 1 из 50 000 000 рождений женского пола). Это может произойти у женщин с больным отцом и матерью-носителем, у тех, у кого отсутствует Х-хромосома, или у тех, у кого есть инактивированная Х-хромосома (наиболее частая из редких причин). Дочь матери-носителя и больного отца будет поражена или станет носителем с равной вероятностью, так как она всегда унаследует пораженную Х-хромосому от своего отца и имеет 50% шанс унаследовать пораженную Х-хромосому от своей матери. .
Было замечено, что нарушение гематоэнцефалического барьера является заметной особенностью развития МДД.
Другие формы заболевания
К менее распространенным видам относятся:
- Дистрофия Эмери-Дрейфуса — Х-сцепленная патология, связанная с мутацией гена Xq28. Болезнь обычно дебютирует в возрасте 4-15 лет с укорочения ахилловых сухожилий, из-за чего ребенок не может опуститься на пятки. Затем возникает слабость мышц плече-лопаточной области, перонеальных мышц, разгибающих стопу, задних затылочных мышц. Возникают контрактуры локтевых суставов, нарушается работа сердца5.
- Миодистрофия Эрба-Рота, вид поясно-конечностной дистрофии мышц, передается по аутосомно-рецессивному типу. Первые признаки появляются в юношеском возрасте — в 10-20 лет. Больные начинают испытывать трудности при беге, быстрой ходьбе, прыжках, вызванные слабостью мышц тазового пояса. По мере прогрессирования меняется осанка, походка, снижается тонус мышц плечевого пояса.
Через 10-20 лет после дебюта дистрофии Эрба-Рота больной может потерять способность ходить6. - Миодистрофия Ландузи-Дежерина начинает проявляться в возрасте 10-20 лет, первыми симптомами часто становятся слабость мимических мышц, лопаток и плеч. Больным сложно поднимать руки над головой, появляются «крыловидные» лопатки и сколиоз. По мере прогрессирования поражаются круговые мышцы рта и глаз, из-за чего сложно крепко зажмурить глаза и сжать губы7.
Лечение миопатии и амиотрофии – наша работа. Клиника “Эхинацея”
Мы предлагаем Вам пройти лечение и обучение самостоятельному выполнению восстановительных процедур. Клиника “Эхинацея” принимает пациентов из любой точки России и мира.
Как проходит лечение и обучение:
- Все начинается с первичного осмотра врачом неврологом, при необходимости выполняем обследование (электромиография, анализы, исследования сердца, и другие, по результатам осмотра).
- По результатам осмотра и обследования мы составляем Вам индивидуальный план лечения на ближайшие 6-12 месяцев (лекарства, питание, гимнастика и массаж, физиолечение, режим нагрузок).
- Затем Ваш лечащий врач и специалист – реабилитолог осматривают Вас совместно и в ходе осмотра уточняют программу физической реабилитации (массаж, гимнастика, физиолечение).
- Сразу же реабилитолог приступает к выполнению программы физической реабилитации. В течение 3-3,5 часов выполняются все необходимые процедуры, в ходе которых реабилитолог обеспечивает и обучение сопровождающего (и/или самого пациента) самостоятельному выполнению процедур по той методике, которая нужна пациенту. Таких занятий может понадобиться от одного до пяти, ежедневно или через день. Если Вы приезжаете из другого города, основную программу обследования, реабилитации и обучения можно пройти за 5 дней, с понедельника по пятницу.
- Если сформировалась деформация позвоночника (сколиоз), выполняется мягкая мануальная терапия, специально подобранная для лечения миопатов; процедура выполняется лечащим врачом лично.
- Если ситуация того требует (депрессия, отсутствие мотивации к поддержанию здоровья), возможно выполнение и более серьезного психотерапевтического лечения, разнообразные разгрузочные и психотерапевтические сеансы в ходе лечения в клинике.
- Через 6-12 месяцев рекомендуем повторить консультацию, короткий курс рабилитационных процедур и получить план лечения на следующие 6-12 месяцев.
По окончании курса Вы будете уметь самостоятельно выполнять необходимые для поддержания своего здоровья процедуры. В ходе лечения улучшается общее самочувствие, повышается сила мышц, начинают возвращаться утраченные двигательные навыки. Вам будут даны подробные инструкции и план лечения на ближайшие 6-12 месяцев, для выполнения его по месту жительства. При необходимости мы примем Вас на повторное лечение через полгода, год или больший промежуток времени.
Если Вы едете к нам из другого города, рекомендуем приезжать на период 5 дней – с понедельника по пятницу. Если Вы не располагаете этим временем, курс можно уложить в 3 дня, хотя и с некоторыми потерями.
Патогенез
Мышечная миопатия Дюшенна развивается на фоне мутации в гене, кодирующем белок дистрофин в локусе Xp21.2. Практически все дети унаследуют дистрофию от матери-носителя дефектного гена. Причем женщины-носители патологически измененного гена не подвержены миопатии Дюшенна, что обусловлено наличием второй половой хромосомы, обеспечивающей выработку дистрофина. Также генная мутация может возникать в материнской яйцеклетке. Тяжелое течение миопатии Дюшенна обусловлено генетическими аберрациями, приводящими к нарушению считывания дезоксирибонуклеиновой кислоты и абсолютному прекращению синтеза дистрофина, влекущее за собой разрушение мышечных клеток (миоцитов).
Мышечная дистрофия
Мышечная дистрофия, группа хронических наследственных заболеваний скелетных (произвольных) мышц человека. Мышечные дистрофии проявляются прогрессирующей слабостью и дегенерацией мышц.
Существует целый ряд форм мышечной дистрофии. Они различаются по таким характеристикам, как возраст, в котором начинается заболевание, локализация пораженных мышц, выраженность мышечной слабости, скорость прогрессирования дистрофии и тип ее наследования. Чаще всего встречаются две формы: мышечная дистрофия Дюшенна и миотоническая мышечная дистрофия.
Мышечная дистрофия Дюшенна
(псевдогипертрофическая мышечная дистрофия) – самая частая форма этого заболевания у детей. Причина болезни – генетический дефект, локализованный на X-хромосоме (одной из двух хромосом, определяющих пол человека).
Женщины с дефектным геном передают его своим детям, но у них самих симптомы дистрофии, как правило, отсутствуют. У мальчиков, получивших дефектный ген, в возрасте от двух до пяти лет неизбежно развивается мышечная слабость.
В первую очередь страдают крупные мышцы нижних конечностей и тазового пояса. Затем дегенерация распространяется на мышцы верхней половины тела, а далее постепенно и на все основные группы мышц.
Характерное проявление болезни – псевдогипертрофия икроножных мышц, т.е. увеличение их за счет отложения жира и разрастания соединительной ткани.
Мышечная дистрофия Дюшенна – одна из самых тяжелых и быстро прогрессирующих форм. К 12 годам больные обычно теряют способность передвигаться, а к 20 годам большинство из них погибает.
Миотоническая мышечная дистрофия
(болезнь Штейнерта) – наиболее распространенная форма мышечной дистрофии у взрослых. Она обусловлена дефектным геном на 19-й хромосоме. Мужчины и женщины страдают в равной степени и могут передать генетический дефект детям.
Заболевание проявляется в любом возрасте, в том числе в младенчестве, но чаще всего – между 20 и 40 годами. Первыми симптомами служат миотония (замедленное расслабление мышц после сокращения) и слабость мимических мышц; возможно также поражение мышц конечностей и других частей тела.
Прогрессирование болезни происходит в большинстве случаев медленно, и полная инвалидность может наступить не ранее чем через 15 лет.
Особенность данного заболевания состоит в том, что помимо произвольных мышц оно поражает также гладкую мускулатуру и сердечную мышцу.
Патоморфология
Все формы мышечной дистрофии характеризуются дегенерацией мышц, но не связанных с ними нервов. В пораженной мышечной ткани обнаруживают различные изменения, в том числе значительные колебания толщины (диаметра) мышечных волокон. Постепенно эти волокна теряют способность сокращаться, распадаются и замещаются жировой и соединительной тканью.
Диагноз
По своим клиническим проявлениям мышечные дистрофии сходны со спинальными амиотрофиями – наследственными болезнями, поражающими двигательные нейроны спинного мозга.
Эти болезни тоже приводят к выраженной мышечной слабости, иногда угрожающей жизни.
Для подтверждения диагноза мышечной дистрофии может потребоваться электромиография, а иногда и биопсия мышц с микроскопическим исследованием для выявления характерных дистрофических изменений.
Причины
Специалисты полагают, что каждая форма мышечной дистрофии обусловлена отдельным точечным генетическим дефектом, нарушающим способность мышечных клеток синтезировать необходимые белки.
Усилия исследователей сосредоточены на поиске дефектов, лежащих в основе заболеваний, и тех отклонений в составе белков, к которым эти дефекты приводят.
В настоящее время выявлен ген мышечной дистрофии Дюшенна.
Лечение
Не существует способов предотвратить или замедлить прогрессирование мышечной слабости при мышечной дистрофии.
Терапия направлена главным образом на борьбу с осложнениями, такими, как деформация позвоночника, развивающаяся вследствие слабости мышц спины, или предрасположенность к пневмониям, обусловленная слабостью дыхательных мышц.
В этом направлении достигнуты определенные успехи, и качество жизни больных с мышечной дистрофией улучшилось. Сейчас многие больные, несмотря на свой недуг, могут вести полнокровную и продуктивную жизнь.
Проверь себя!
Ответь на вопросы викторины «Фобии и страхи»
Чего больше всего боятся трискаидекафобы?
Этиология возникновения паралича Дюшена-Эрба
Как было указано ранее, чаще всего встречается верхний акушерский паралич, развивающийся при родовом травмировании плода. В иных возрастных категориях заболевание может быть вызвано повреждением плечевых спинномозговых корешков, находящихся в области 5 и 6 позвонков. Повредить нервные волокна можно при сильном верхнем ударе, падении на сторону руки, ушибе об статичный предмет, путем «выдергивания» руки в сторону, проникающем ранении в указанную зону.
Детский паралич образуется при растяжении нервного ствола плечевого сплетения во время поворота за ногу, вытягивания плода за руки, освобождение плеча из родовых путей, вытяжении за таз. Описанные манипуляции необходимы при неправильном предлежании ребенка, недостаточно интенсивной родовой деятельности, затяжном периоде родоразрешения, узкой анатомии таза у матери, крупном плоде. Часто вместе с приобретенным параличом имеет место сочетание с кривошеей.
Первичными проявлениями полученной травмы является воспаление, гематома на поврежденном участке. Происходит надрыв или полный разрыв нервных волокон, сосудистых стенок, мускулатуры. Со временем острые проявления исчезают, однако, на месте нарушения целостности тканей образуются рубцы, которые давят на нервное сплетение и питающие отдел сосуды.
Благотворительные фонды помогают приехать на лечение в Израиль
Международный отдел медицинского центра Хадасса работает в тесном контакте с ведущими благотворительными фондами, которые уже обеспечили десятки больных редкими заболеваниями детей средствами, необходимыми для прохождения лечения в МЦ Хадасса. Мы настоятельно советуем всем родителям, которые взвешивают возможность привести своих детей на лечение в Израиль, обратиться в эти организации.
- Русфонд
- World Vita
- Благотворительный фонд «Алеша»
- Благотворительный фонд «Артемка»
- Благотворительный фонд «Клуб добряков»
- Благотворительный фонд «Помогать легко»
У вас возникли вопросы? Обращайтесь к нам и получите ответ
по телефону: +972 2 560-97-99 (круглосуточно)
по электронной почте: ru-office@hadassah.org.il
или заполнив контактную форму
Патологическая анатомия
Морфологические изменения при Миопатии характеризуются нарастающей атрофией скелетных мышц, к-рые уменьшаются в объеме и становятся плотными, бурого цвета вследствие разрастания соединительной ткани или, напротив, увеличиваются в объеме за счет жировой клетчатки.
Рис. 1. Микропрепарат мышцы (поперечный срез) при миопатии Дюшенна в стадии частичной сохранности двигательной функции: беспорядочное расположение разнокалиберных мышечных волокон — атрофированных (1), нормального диаметра (2), единичных гипертрофированных (3); дистрофические изменения в части мышечных волокон (4); разрастание соединительной ткани в эндомизии (5); гематоксилин-эозин; X 200.
Рис. 2. Микропрепарат мышцы (продольный срез) при миопатии Дюшенна в стадии обездвиженности: единичные атрофированные мышечные волокна (1) среди фиброзной и жировой ткани; лимфоидно-гистиоцитарная инфильтрация (2); гематоксилин-эозин; X 200.
При различных формах мышечных дистрофий (псевдогипертрофических, плече-лопаточно-лицевой, тазоплечевой, офтальмоплегической, бульбарно-офтальмоплегической) определяются в основном однотипные гистол, изменения (рис. 1): уменьшение количества мышечных волокон в пучках, резкая диффузная разнокалиберность сохранившихся волокон с преобладанием среди них атрофированных, гиалиновая и вакуольная дистрофия в части мышечных волокон, дискоидный и коагуляционный некроз отдельных волокон, расщепление гипертрофированных волокон, разрастание соединительной и жировой ткани в эндо- и перимизии. В нек-рых мышечных волокнах находят саркоплазматические тельца, саркоплазматические массы, кольцевидные миофибриллы. Изредка встречаются регенерирующие волокна с круглыми сочными ядрами и богатой рибонуклеопротеидами саркоплазмой. Мышечные веретена длительное время остаются неизмененными. В части наблюдений выявляются периваскулярные лимфоидно-гистиоцитарные инфильтраты. По мере нарастания двигательных нарушений отмечается постепенное уменьшение количества мышечных волокон и нивелировка их диаметра за счет резкого уменьшения количества и калибра гипертрофированных волокон. Наиболее быстро эти изменения развиваются при миопатии Дюшенна — злокачественном варианте псевдогипертрофической М. Для доброкачественного варианта — миопатии Беккера — характерно сочетание выраженных процессов липоматоза н склероза с резкой гипертрофией части мышечных волокон. С последней особенностью связывают длительную компенсацию двигательных нарушений у таких больных. В далеко зашедшей стадии М. определяются небольшие островки из атрофированных мышечных волокон на фоне резкого склероза и липоматоза эндо- и перимизия (рис. 2), при этом гистологически не представляется возможным дифференцировать М. с нейромышечной атрофией. У пробандов при биопсии мышц находят единичные атрофированные мышечные волокна преимущественно I типа, пролиферацию ядер с переходом их в центр волокна и незначительное увеличение соединительной ткани в эндомизии.
Наиболее ранние ультраструктурные изменения в мышцах при Миопатии характеризуются утолщением и расщеплением Z-линий с последующим разрушением миофибрилл мышечного волокна. Благодаря фазово-контрастной и электронной микроскопии выявлены дефекты в мембранных системах мышечного волокна. Гистоферменто химически отмечается ослабление реципрокных отношений между гликолитическими и окислительными ферментами в мышечных волокнах различного типа.
В центральной и периферической нервной системе изменений не обнаружено. Характерен кардиосклероз.
Как диагностировать заболевание?
 Предварительный сбор анамнеза, наличие отметок в истории болезни ребенка о сложных родах, нахождение руки в постоянном вытянутом положении помогает специалистам поставить корректный диагноз. Заболевание во взрослом возрасте требует дифференциации от иных патологий со схожей симптоматикой.
Предварительный сбор анамнеза, наличие отметок в истории болезни ребенка о сложных родах, нахождение руки в постоянном вытянутом положении помогает специалистам поставить корректный диагноз. Заболевание во взрослом возрасте требует дифференциации от иных патологий со схожей симптоматикой.
Постановкой диагноза занимаются врачи-неврологи и ортопеды. Врожденное расстройство диагностируется неонатологом в первые дни жизни младенца. К аппаратным методам диагностирования заболевания относятся:
- Рентгенографическое исследование. Помогает обнаружить изменения, происходящие в костях изучаемой зоны. В первые месяцы жизни маленького пациента просматривается расхождение промежутка внутрисуставной области. Могут быть выявлены признаки атрофии костной ткани.
- Ультразвуковое исследование. Применяется в качестве подтверждающего способа.
- Миелография.
- Электронейрография. Оценивается интенсивность прохождения импульса по стволу нервного канала.
- Магнитно-резонансная томография. Используется как сопутствующая диагностика, позволяющая оценить состояние мягких тканей.
- Компьютерное сканирование. Самый информативный способ при планируемой операции. Одинаково эффективно визуализирует как костные образования, так и нервные пути.
Томографическое аппаратное обследование можно провести только в мед-организации, обладающей необходимым оборудованием и опытными функциональными диагностами. Не каждая районная поликлиника способна предоставить соответствующие услуги. Для быстрого поиска диагностического центра МРТ и КТ на нашем портале представлен перечень рейтинговых клиник, расположенных в каждом районе столицы. Пользователи портала могут изучить подробную контактную информацию, сравнить цены на диагностические услуги, бесплатно проконсультироваться и записаться на процедуру сканирования по телефону горячей линии, расположенному в верхней части страницы.
Лекарства при миодистрофии Дюшенна
Медикаментозное лечение является важной частью общей терапии. Наиболее популярной группой препаратов для пациентов с миодистрофией Дюшенна являются стероиды: преднизолон, дефлазакорт и другие
Начинают их прием еще в возрасте 4-6 лет, когда проявляются признаки болезни, но существенного ухудшения состояния нет. Стероиды помогают отсрочить развитие тяжелых симптомов, но в долгосрочной перспективе они неэффективны. Как только у пациента начинает прогрессировать атрофия, улучшить состояние мышц эти лекарства уже не могут. И все же стероиды остаются важной составляющей лечения дистрофии Дюшенна.
Существуют и другие разработки, направленные на улучшение работы мышц. Так, в 2016 году FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) одобрило лекарство Exondys51 (этиплирсен), которое сможет стать конкурентом стероидам. По данным проведенных исследований, вещество способно укрепить мышечную ткань, ведь оно увеличивает уровень дистрофина в скелетных мышцах. Однако не все врачи разделяют мнение FDA, поскольку утверждение препарата проходило по ускоренной процедуре. По факту сейчас нет однозначных доказательств, что он может отсрочить развитие паралича или ослабить симптомы развитого заболевания. А значит, Exondys51, как и стероиды, проявляет эффективность лишь в коротком периоде болезни. Несмотря на то, что лекарство уже утверждено, FDA продолжает исследования. Если эффективность препарата не будет подтверждена, его производство остановят.
Европейский Комитет по лекарственным препаратам для человека (CHMP) в 2014 году зарегистрировал другой медикамент — Трансларна (аталурен). В отличие от других лекарств, которые только укрепляют мышцы, аталурен способен восстанавливать нарушенный синтез белков. А значит, теоретически устраняет саму причину развития болезни. Поскольку это новая разработка, понять, насколько она эффективна в долгосрочной перспективе, пока трудно. Но промежуточные результаты достаточно обнадеживающие.