Хромосомные нарушения
Содержание:
- Классификация мутаций
- Literature
- Амплификации, делеции генов и хромосомные аберрации в опухоли
- Известные заболевания хромосомной природы
- Скрининг на генетические заболевания
- Структурные аномалии хромосом
- Этиология хромосомных заболеваний
- Причины
- Генетическое исследование и аутосомно-доминантные заболевания
- Как это касается других членов семьи
- Флуоресцентная гибридизация in situ (FISH)
- Хромосомные аберрации в опухолях солидного типа
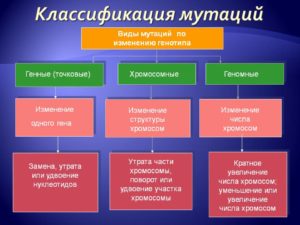
Классификация мутаций
-
По характеру проявления в гетерозиготном состоянии – доминантные (проявляются в гетерозиготном состоянии) и рецессивные (проявляются только в гомозиготном состоянии).
-
В зависимости от причины – спонтанные (без видимых причин) и индуцированные (вызванные направленным действием какого-то фактора).
-
В зависимости от локализации в клетке – ядерные и цитоплазматические.
-
По отношению к возможности наследования – генеративные (в половой клетке) и соматические (возникшие в соматической телесной клетке). Соматические мутации у видов, размножающихся половым способом, по наследству не передаются. Но для данного индивида они не безразличны (например, родимые пятна, пятна на радужке, раковая опухоль).
-
Функциональная (в зависимости от исхода) – полезные, вредные (в том числе летальные) и нейтральные (безразличные).
-
По характеру изменения генома – генные (изменение структуры гена), хромосомные (изменение строения хромосом)и геномные (изменение числа хромосом).
Генные мутации
В основегенных мутаций лежит изменение в строениимолекулы ДНК. Все они могут быть объединеныв три группы.
-
Замена одних азотистых оснований на другие. Например, при дезаминировании (цитозин превращается в тимин) или при ошибочном включении нуклеотида в процессе репликации ДНК.
-
Сдвиг рамки считывания – в результате выпадения или вставки какого-то нуклеотида в синтезируемую цепь.
ААА ЦГТ ААЦ фен – ала – лей
ААА АЦГ ТАА фен – цис – иле
кодогенная цепьДНК полипептид
-
Изменение порядка нуклеотидов в пределах гена (при повороте на 1800 участка цепи ДНК).
Хромосомные мутации
Воснове хромосомных мутаций лежатизменения в строении хромосом. Ониподразделяются на внутри-и межхромосомные.
а)дефишенси– отрывконцевого участка хромосомы;
б)делеция– выпадениесрединного участка хромосомы;
в)дупликация– удвоениеучастка хромосомы;
г)инверсия– поворотучастка хромосомы на 180о.Инверсия может быть перицентрической(захватывает центромеру) и парацентрической(в пределах одного какого-то плеча).
а)транслокация– в основележит отрыв участка одной хромосомы иприсоединение его к другой хромосоме.Разновидности транслокаций: реципрокная(взаимный обмен плечами) и робертсоновская– центрическое разделение или слияниеотдельных хромосом.
Предполагают, чтов процессе превращения обезьяны(шимпанзе) в человека имело место слияниедвух акроцентрических хромосом в однуметацентрическую.
б)транспозиция– перемещениенебольших участков генетическогоматериала в пределах как одной хромосомы,так и всего кариотипа.
Геномные мутации
Воснове лежит изменение числа хромосом.Различают два вида таких мутаций:
- полиплоидия – увеличение числа хромосом на величину, кратную гаплоидному набору;
- анеуплоидия– увеличение числа хромосом на величину, не кратную гаплоидному набору. В случае трисомии имеется одна лишняя хромосома (набор 2n + 1), при моносомии одна хромосома отсутствует (набор 2n – 1), при нулисомии отсутствует целиком хромосомная пара (2n – 2).
Полиплоидияшироко распространена в растительноммире. Так, существует три вида пшеницы(2n, 4n,6n),где n= 7 . Хризантемыимеют наборы от 2nдо22n(n= 9).
Аналогичныепримеры можно найти у всех растений,как дикорастущих, так и культивируемых.Поэтому считается, что эволюция растений шла по пути полиплоидизации.
Полиплоидияшироко используется в селекционнойработе (у полиплоидных растений крупнееплоды, больше семян).
Учеловека установлено рождение триплоидов,однако они нежизнеспособны (существуютот нескольких минут до несколькихчасов).
Геномныеи хромосомные мутации у человека лежатв основе группы заболеваний,которые были названы хромосомнымиболезнями.
Literature
- Baranov V. S., Kuznecova T. V. Citogenetika jembrional’nogo razvitija cheloveka: nauchno-prakticheskie
aspekty // SPb.: Nauchnaja literatura, 2007. — S. 252—310. - Vorsanova S. V., Jurov Ju. G., Chernyshov V. N. Medicinskaja citogenetika // Moskva, 2006. — S. 219—222.
- Biancotti J. C. Human embryonic stem cells as models for aneuploid chromosomal syndromes // Stem cells. — 2010. — Vol. 28. — P. 1530—1540.
- Bricker L. Types of pregnancy loss in recurrent miscarriage: implications for research and clinical practice // Hum. Reprod., 2002. — Vol. 17, N 5. — P. 1345—1350.
- Carrell D. T. The clinical implementation of sperm chromosome aneuploidy testing: pitfalls and promises // J Androl.,2008. — Vol. 29(2). — P. 124—33.
- Gersen S. L. The principles of clinical Cytogenetics // New York: Springer, 2013. — P. 275—292
- Harper J. C. Preimplantation genetic diagnosis // CambridgeUniversity Press, 2009. — Р. 95—116.
- Harton G. L. at al. ESHRE PGD Consortium/Embryology Special Interest Group. Best practice guidelines for polar body and embryo biopsy for preimplantation genetic diagnosis/screening (PGD/PGS) // Human Reproduction, 2010. — Vol. 1. — P. 1—8.
- Nielsen J. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark // Hum Genet., 1991. — Vol. 87(1). — P. 81—3.
- McKinlay Gardner R. J. Chromosome abnormalities and genetic counseling // New York: Oxford University Press, Inc., 2012, — P. 27—66, P. 377—402.
Библиографическая ссылка
Гонтарь Ю. В., Ильин И. Е., Будерацкая Н. А., Связь количественных хромосомных аномалий сперматозоидов и преимплантационных эмбрионов // «Живые и биокосные системы». — 2014. — № 8; URL: http://www.jbks.ru/archive/issue-8/article-11.
Амплификации, делеции генов и хромосомные аберрации в опухоли
Хромотрипсис представляет собой один из вариантов комплексных изменений в геноме, который приводит к множественным перестройкам в хромосомах. Это явление было открыто относительно недавно, механизм до конца не изучен. Сама суть процесса заключается в том, что хромосома распадается на множество мелких фрагментов, которые затем собираются хаотично. При этом некоторые участки могут быть безвозвратно утеряны. Явление хромотрипсиса не является массовым, оно присуще только единичным хромосомам.
Еще одним вариантом хромосомных аберраций в опухоли, который был открыт недавно и в настоящее время активно изучается, является геномная нестабильность. Суть данного явления заключается в том, что опухолевые клетки, относящиеся к одному и тому же гистологическому типу, содержат различные варианты мутаций. Именно поэтому злокачественные новообразования демонстрируют большое разнообразие повреждений генома, что приводит к разнообразию вариантов роста и метастазирования.
Известные заболевания хромосомной природы
Одним из самых известных заболеваний, происходящих по причине наличия аномалий в генетическом материале, является синдром Дауна. Он обуславливается трисомией по 21 хромосоме. Характерным признаком этой болезни является отставание в развитии. Дети испытывают серьезные проблемы во время обучения в школе, часто им требуется альтернативная методика преподавания материала. Вместе с тем отмечаются нарушения физического развития – плоское лицо, увеличенные глаза, клинодактилия и другие. Если такие люди прикладывают значительные усилия, они могут достаточно хорошо социализироваться, известен даже случай успешного получения высшего образования мужчиной с синдромом Дауна. У больных повышен риск заболеть деменцией. Это и ряд других причин приводит к небольшой продолжительности жизни.
К трисомии относится и синдром Патау, только в этом случае имеется три копии 13 хромосомы. Для заболевания характерны множественные пороки развития, часто с полидактилией. В большинстве случаев отмечается нарушение деятельности центральной нервной системы либо ее неразвитость. Часто (примерно в 80 процентах) больные имеют пороки развития сердца. Тяжелые нарушения приводят к высокой смертности – в первый год жизни умирает до 95% детей с этим диагнозом. Заболевание не поддается лечению или коррекции, как правило, можно лишь обеспечить достаточно постоянный контроль состояния человека.
Еще одна форма трисомии, с которой рождаются дети, относится к 18 хромосоме. Заболевание в этом случае носит название синдрома Эдвардса и характеризуется множественными нарушениями. Деформируются кости, часто наблюдается измененная форма черепа. Сердечно-сосудистая система обычно с пороками развития, также проблемы отмечаются с органами дыхания. В результате около 60% детей не доживают до 3 месяцев, к 1 году умирает до 95% детей с этим диагнозом.
Трисомия по другим хромосомам у новорожденных практически не встречается, поскольку почти всегда приводит к преждевременному прерыванию беременности. В части случаев рождается мертвый ребенок.
С нарушениями числа половых хромосом связан синдром Шерешевского-Тернера. Из-за нарушений в процессе расхождения хромосом теряется X-хромосома в женском организме. В результате организм не получает должного количества гормонов, поэтому нарушается его развитие. В первую очередь это относится к половым органам, которые развиваются лишь отчасти. Практически всегда для женщины это обозначает невозможность иметь детей.
У мужчин полисомия по Y или X хромосоме приводит к развитию синдрома Клайнфельтера. Для этого заболевания характерна слабая выраженность мужских признаков. Зачастую сопровождается гинекомастией, возможно отставание в развитии. В большинстве случаев наблюдаются ранние проблемы с потенцией и бесплодие. В этом случае, как и для синдрома Шерешевского-Тернера, выходом может стать экстракорпоральное оплодотворение.
Скрининг на генетические заболевания
Сегодня известно более 11 000 моногенных заболеваний, которые кодируются одним геном (генетически обусловленные) и передаются от родителей их потомкам. Механизм передачи многих генетических болезней объясняется принципами Менделя.
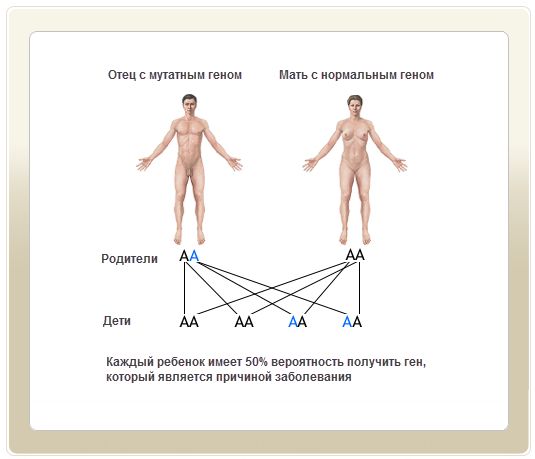
Аутосомно-доминантные моногенные синдромы встречаются с частотой 1: 200 индивидов; заболевание наблюдается у многих поколений, передается потомкам и рецидивирует с частотой 50%. Примерами аутосомно-доминантных моногенных расстройств могут быть:
- ахондроплазия,
- нейрофиброматоз,
- синдром Марфана,
- болезнь Хантингтона,
- семейный полипоз.
Появление аутосомно-доминантных заболеваний у новорожденных от «здоровых» родителей может быть обусловлено следующими причинами:
1. Мозаицизм зародышевых клеток. Мутация может иметь место лишь в популяции зародышевых клеток. Итак, родители являются непораженными, но могут передавать мутацию потомкам.
2. Новые мутации. Рост возраста родителей ассоциируется с увеличением риска аутосомно-доминантных расстройств (ахондроплазии, танатофорной дисплазии, нейрофиброматоза, синдрома Аперта — краниосиностоз). Риск рецидивов у других детей не увеличивается.
3. Вариабельна экспрессия. Тяжесть заболевания может варьировать, и родители могут не распознать мягкие и субклинические мутации.
4. Уменьшенная пенетрантность. Родители могут иметь аномальный ген без клинических проявлений заболевания.
5. Неверное отцовство. Частота неверного отцовства достигает 15%.
Аутосомно-рецессивные моногенные заболевания проявляются в многочисленных родственников при наличии двух пораженных аллелей. Если оба родителя являются носителями пораженного гена, риск заболевания у потомства равен 25% при каждой беременности. Аутосомно-рецессивные заболевания включают кистозный фиброз, серповидно-клеточную анемию, фенилкетонурию, болезнь Тея-Сакса, Канавана и др.
При Х-сцепленных рецессивных синдромах (гемофилия и др.) мать-носитель пораженного гена передает его своим сыновьям. Итак, 50% сыновей могут быть больными и 50% дочерей будут носителями этого гена. Редкие Х-доминантные синдромы могут передаваться от каждого родителя каждому ребенку подобно аутосомно-доминантных синдромов. Фенотип может сильно варьировать, что связано со смешанной пенетрантностью, лионизацией (гетерохроматизацией) Х-хромосомы (синдром ломкой Х-хромосомы) и геномным импринтингом.
Экспансия тринуклеотидных повторов. Некоторые гены содержат участки тройных повторов (например, ССС). Такие участки являются нестабильными и могут увеличиваться в следующих генерациях, этот феномен получил название антиципации. Количество повторений определяет степень поражения индивида. Экспансия тринуклеотидных повторов составляет основу многочисленных генетических расстройств, таких как синдром ломкой (фрагильной) Х-хромосомы, миотоническая дистрофия и болезнь Хантингтона.
Синдром ломкой (фрагильной) Х-хромосомы является наиболее частой причиной семейной задержки умственного развития. Пораженные мужчины имеют типичные черты: большие уши, выступающая челюсть, большие яички, аутичное поведение, легкая или умеренная умственная отсталость. Женщины обычно менее поражены в связи с инактивацией Х-хромосомы.
Ген ломкой Х-хромосомы локализуется в Х-хромосоме и имеет три нуклеотидные повтора (ССС). Нормальные индивиды имеют 6-50 повторов, непораженные носители женского пола могут иметь 50-200 повторов, которые могут распространяться на мейоза до полной мутации при наличии более 200 повторов. Если имеет место полная мутация, ген инактивируется путем метилирования; плод будет пораженным. Тяжесть заболевания зависит от степени Х-инактивации у женщин, степени метилирования и мозаицизма размера повторов.
Женщины-носители премутации имеют 50%-й риск передачи гена с экспансией. Мужчины с премутациею фенотипически являются нормальными, но все их дочери будут носителями премутации. В случае трансмиссии мужчинам количество повторов остается стабильным. Тест на ломку Х-хромосому выполняется с целью выявления количества повторов и степени метилирования.
Структурные аномалии хромосом
Структурные аномалии хромосом могут касаться одной или более хромосом и обычно возникают через разрывы хромосом. Разрывы хромосом могут быть вызваны действием агрессивных факторов внешней среды: вирусов, радиации или химических веществ. Последствия разрыва хромосом зависят от дальнейшей судьбы разорванных частиц. В некоторых случаях оторвана часть хромосомы теряется — частичная делеция хромосомы. В случаях частичной делеции хромосомы ребенок характеризуется определенными аномалиями.
Синдром «кошачьего крика» обусловлен частичной делецией короткого плеча хромосомы 5 и характеризуется микроцефалией, умственной отсталостью, врожденными пороками сердца. Плач таких детей напоминает кошачий крик.
Микроделеции хромосом охватывают лишь несколько смежных генов и могут приводить к развитию синдрома микроделеции, или синдрома смежных генов. Места возникновения таких делеций называют смежными генными комплексами. Их идентифицируют с помощью высокотехнологичных дифференциальной окраски хромосомных полос.
Примером может быть микроделеция длинного плеча хромосомы 15 (15 ^ 11-15 ^ 13). При наследовании пораженной делецией материнской хромосомы возникает синдром Ангельмана, при котором ребенку присуща умственная отсталость, задержка речевого и моторного развития, непровоцированные приступы смеха. При наследовании дефектной родительской хромосомы возникает синдром Прадера-Вилли. Пораженные индивиды страдают от гипотонии, ожирения, умственной отсталости, гипогонадизма и крипторхизма. Такое различие экспрессии генетического материала в зависимости от наследования родительской или материнской хромосомы является примером геномного импринтинга.
Другими генными синдромами, которые могут наследоваться от обоих родителей, является синдром Миллера-Дикер в результате делеции 17р 13 (задержка развития, врожденные пороки лица и сердца) и синдром Шпринтцена при делеции 22 ^ 11 (дефекты неба, пороки развития сердца, задержка речевого развития, нарушение способности к обучению, шизофреноподобные расстройства).
Ломкие места — это участки хромосом, проявляющих склонность к отрыву или разрушения при определенных манипуляциях с клеткой. Ломкие места можно обнаружить при культивировании лимфоцитов в среде с дефицитом фолатов. Синдром ломкой Х-хромосомы сопровождается умственной отсталостью, большими ушами, выступание челюсти и бледными голубыми радужками.
Мужчины поражаются чаще, чем женщины (4/2000 сравнению с 1/2000), чем объясняется преобладание мужского пола среди умственно отсталых лиц. Синдром ломкой Х-хромосомы занимает второе место после синдрома Дауна среди причин умственной отсталости хромосомного происхождения.
Этиология хромосомных заболеваний
К этиологическим факторам хромосомных патологий относятся все разновидности хромосомных мутаций. Кроме того, некоторые геномные мутации также способны оказывать подобное действие.
У человека встречаются делеции, дупликации, транслокации и инверсии, то есть все типы мутаций. При делеции и дупликации генетическая информация оказывается в недостаточном и избыточном количестве соответственно. Поскольку современными методами можно выявить отсутствие даже небольшой части генетического материала (на уровне гена), то провести четкую границу между генными и хромосомными заболеваниями практически невозможно.
Транслокации представляют собой обмен генетическим материалом, который происходит между отдельными хромосомами. Иными словами, происходит перемещение участка генетической последовательности на негомологичную хромосому. Среди транслокаций выделяют две важные группы – реципрокные и Робертсоновские.
Транслокации реципрокного характера без потери задействованных участков называются сбалансированными. Они, как и инверсии, не вызывают потери генной информации, поэтому не приводят к паталогическим эффектам. Тем не менее, при дальнейшем участии таких хромосом в процессе кроссинговера и редукции могут образовываться гаметы с несбалансированными наборами, обладающие недостаточным набором генов. Их участие в процессе оплодотворения приводит к тому, что у потомства развиваются те или иные наследственные синдромы.
Для Робертсоновских транслокаций характерно участие двух акроцентрических хромосом. В ходе процесса короткие плечи утрачиваются, а длинные сохраняются. Из 2 исходных хромосом формируется одна цельная, метацентрическая. Несмотря на потерю части генетического материала развития патологий в таком случае обычно не происходит, поскольку функции утраченных участков компенсируются аналогичными генами в остальных 8 акроцентрических хромосомах.
При концевых делециях (то есть при их утрате) может сформироваться кольцевая хромосома. У ее носителя, получившего такой генный материал от одного из родителей, отмечают частичную моносомию по концевым участкам. При разрыве через центромеру может сформироваться изохромосома, имеющая одинаковые по набору генов плечи (у обычной хромосомы они отличаются).
В некоторых случаях может развиваться однородительская дисомия. Она возникает, если при нерасхождении хромосом и оплодотворении возникнет трисомия, а после этого одна из трех хромосом будет удалена. Механизм этого явления в настоящее время не изучен. Однако в результате в хромосомном наборе появится две копии хромосомы одного родителя, в то время как часть генной информации от второго родителя будет утеряна.
Многообразие вариантов искажения хромосомного набора обуславливает различные формы заболеваний.
Причины
Структурные изменения, происходящие в хромосомах, являются следствием нарушения их целостности в результате разрывов парных нитей спирали ДНК. Этот процесс может происходить самопроизвольно, но чаще его провоцирует воздействие внешних факторов (мутагенов). Спонтанные перестройки случаются очень редко (примерно 1-100 случаев на 1 млн. представителей вида) и тоже подчиняются мутационному давлению окружающей среды, но ввиду неуточненной природы их возникновения, их принято относить к неиндуцированным.
Точные причины хромосомных мутаций в каждом отдельном случае установить тяжело, но выявлена связь между факторами внешней среды и возникновением индуцированных аберраций. Факт установления взаимозависимости внешнего воздействия и мутаций не способствует соотнесению определенных мутагенов с видами перестроек (распределение генов по геному в результате мутаций происходит случайно). Под влиянием одного и того же фактора могут происходить неодинаковые изменения. Все выявленные мутагены подразделяются на 3 вида:
- биологические – бактерии, вирусы, способные встраиваться в ДНК клеток, захватывать часть генов при размножении и переносить их в организм другого человека;
- химические – соединения химического происхождения, большинство из которых созданы искусственным образом (формальдегиды, иприт, нитраты, пестициды, хлороформ, соединения тяжелых металлов, окись азота, консерванты, растворители, красители и др.);
- физические – все виды излучений (радиоактивное, электромагнитное, ультрафиолетовое, нейтронное), высокоэнергетические частицы (альфа- и бета-частицы), слишком высокая и низкая температура, давление.

Генетическое исследование и аутосомно-доминантные заболевания
Из-за многообразия форм заболеваний и наличия сходных по симптомам болезней диагностика классическими методами не всегда оказывается эффективной. Точнее сказать, установленные ей диагнозы в некоторых случаях оказываются ошибочными. Достоверно подтвердить генетический характер заболевания можно только путем анализа наследственного материала. Для этого пациенту потребуется предоставить материал для проведения генетической диагностики (обычно это просто проба крови).
После этого проводится специальное исследование, позволяющее проанализировать наличие в генотипе человека указанных нарушений генетического материала. В связи с большим объемом исследуемой информации проверка может занять длительное время. Кроме того, следует учитывать тот факт, что в ряде случаев результаты теста не являются однозначным указанием на наличие того или иного наследственного заболевания.
По этой причине самостоятельный анализ результатов тестирования едва ли возможен. Для получения достоверной информации рекомендуется обращаться к специалистам по генетическому консультированию. Это врачи с соответствующим образованием, специализирующиеся на работе с наследственным материалом, обладающие достаточным опытом и умеющие рассчитывать вероятность возникновения генетических патологий на основании данных о родителях. Кроме того, генетический консультант поможет правильно настроиться на процесс тестирования, поскольку получение результатов может стать тяжелым испытанием для человека.
В любом случае нужно понимать, что полная и достоверная информация о состоянии здоровья может оказаться незаменимой. Кроме того, пренатальное исследование позволяет заранее убедиться, что на свет появится полноценный малыш без аномалий в генетическом материале.
Как это касается других членов семьи
Если у одного из членов семьи обнаружена хромосомная перестройка, возможно, Вы захотите обсудить этот вопрос с
другими членами семьи. Это даст возможность другим родственникам, при желании, пройти обследование (анализ
хромосом в клетках крови) для определения носительства хромосомной перестройки
Это может быть особенно важно для
родственников, уже имеющих детей или планирующих беременность. Если они не являются носителями хромосомной
перестройки, они не могут передать ее своим детям
Если же они являются носителями, то им могут предложить пройти
обследование во время беременности для анализа хромосом плода.
Некоторым людям сложно обсуждать проблемы, связанные с хромосомной перестройкой, с членами семьи. Они могут
бояться причинить беспокойство членам семьи. В некоторых семьях люди из-за этого испытывают сложности в общении и
теряют взаимопонимание с родственниками. Врачи-генетики, как правило, имеют большой опыт в решении подобных
семейных ситуаций и могут помочь Вам в обсуждении проблемы с другими членами семьи.
Флуоресцентная гибридизация in situ (FISH)
Аббревиатура FISH расшифровывается как fluorescent in situ hybridization – флуоресцентная гибридизация на месте. Это цитогенетический метод, который применяют для выявления и определения положения специфической последовательности ДНК на хромосомах. Для этого используют специальные зонды — нуклеозиды, соединенные с флуорофорами или некоторыми другими метками. Визуализацию связавшихся ДНК-зондов проводят при помощи флуоресцентного микроскопа.
Метод FISH позволяет изучать небольшие хромосомные перестройки, которые не идентифицируются при стандартном исследовании кариотипа. Однако, имеет один существенный недостаток. Зонды являются специфичными только к одному участку генома и, как следствие, при одном исследовании можно определить наличие или число копий только этого участка (или нескольких при использовании многоцветных зондов). Поэтому важным является правильная клиническая предпосылка, а FISH анализ может только подтвердить иди не подтвердить диагноз.
Альтернативой этому методу является хромосомный микроматричный анализ, который при такой же точности, чувствительности и специфичности определяет количество генетического материала в сотнях тысяч (и даже миллионах) точек генома, что дает возможность диагностики практически всех известных микроделеционных и микродупликационных сииндромов.
Хромосомные аберрации в опухолях солидного типа
Солидные опухоли состоят из большого числа недифференцированных клеток, которые склонны к активному делению. Данный тип рака развивается из эпителиальной ткани и характеризуется высокой агрессивностью. Среди примеров вариаций числа копий генов в опухоли солидного строения можно отметить:
- амплификацию гена ERBB2 (он же Her-2/neu). Данный вид мутации имеет большое значение в развитии рака молочной железы и влияет на тактику лечения данного заболевания. Амплификация ERBB2 выявляется примерно у каждой третьей пациентки;
- транслокацию ROS1 и EML4-ALK. В большинстве случаев возникает при немелкоклеточном раке легкого. Выявление данного вида хромосомной аберрации в опухоли оказывает влияние на назначение таргетной терапии, в частности, препарата «Кризотиниб»;
- транслокацию гена RET. Этот ген отвечает за кодирование белка, относящегося к классу рецепторных тирозинкиназ. Транслокации в нем выявляются при эндокринных неоплазиях, раке щитовидной железы, феохромоцитоме, немелкоклеточном раке легкого и других опухолях. Как и в предыдущем случае, диагностика транслокации гена RET позволяет определиться с выбором препарата для таргетной терапии;
- хромосомные аберрации при опухолях головного мозга. Могут выявляться в генах семейства EGFR, VEGFR, PDGFR, C-KIT, BRAF и т.д. Выявление подобных изменений позволяет определить генетический подтип опухоли головного мозга, спрогнозировать клиническое течение, ответ на химиолучевую терапию и определить чувствительность к специфичным лекарственным препаратам.
Выявление описанных выше вариаций числа копий в опухоли солидного типа имеет важное диагностическое значение, поскольку позволяет определиться с тактикой лечения. По этой причине подобные исследования все чаще входят в программу обследования онкологических пациентов