46
Содержание:
Аберрации
При синдроме Дауна существует три копии хромосомы 21.
Хромосомные аберрации — это нарушения нормального хромосомного содержимого клетки и основная причина генетических состояний у людей, таких как синдром Дауна , хотя большинство аберраций практически не имеют эффекта. Некоторые хромосомные аномалии, такие как транслокации или хромосомные инверсии , не вызывают заболевания у носителей, хотя они могут повысить вероятность вынашивания ребенка с хромосомным заболеванием. Аномальное количество хромосом или наборов хромосом, называемое анеуплоидией , может привести к летальному исходу или вызвать генетические нарушения. Генетическое консультирование предлагается семьям, в которых может быть хромосомная перестройка.
Получение или потеря ДНК хромосом может привести к множеству генетических нарушений . Примеры людей включают:
- Cri du chat , вызванная удалением части короткого плеча хромосомы 5. «Cri du chat» на французском языке означает «крик кошки»; заболевание было названо так потому, что пораженные дети издают пронзительные крики, похожие на кошачьи. Больные имеют широко расставленные глаза, небольшую голову и челюсть, умеренные или серьезные проблемы с психическим здоровьем и очень короткие.
- Синдром Дауна , наиболее распространенная трисомия, обычно вызванная дополнительной копией хромосомы 21 ( трисомия 21 ). Характеристики включают снижение мышечного тонуса, более коренастое телосложение, асимметричный череп, раскосые глаза и нарушение развития от легкой до средней степени.
- Синдром Эдвардса , или трисомия-18, вторая по частоте трисомия. Симптомы включают задержку моторики, нарушение развития и многочисленные врожденные аномалии, вызывающие серьезные проблемы со здоровьем. Девяносто процентов заболевших умирают в младенчестве. У них характерные сжатые руки и пальцы внахлест.
- Изодицентрический 15 , также называемый idic (15), частичная тетрасомия 15q или инвертированная дупликация 15 (inv dup 15).
- Синдром Якобсена , который встречается очень редко. Это также называется терминальным нарушением делеции 11q. Пострадавшие имеют нормальный интеллект или легкие отклонения в развитии, с плохими навыками выразительной речи. У большинства из них есть нарушение свертываемости крови, называемое синдромом Пари-Труссо .
- Синдром Клайнфельтера (XXY). Мужчины с синдромом Клайнфельтера обычно бесплодны, обычно выше и имеют более длинные руки и ноги, чем их сверстники. Мальчики с этим синдромом часто застенчивы и тихи, чаще страдают задержкой речи и дислексией . Без лечения тестостероном у некоторых может развиться гинекомастия в период полового созревания.
- Синдром Патау , также называемый D-синдромом или трисомией-13. Симптомы несколько схожи с симптомами трисомии-18, но без характерной сложенной руки.
- Маленькая дополнительная маркерная хромосома . Это означает, что есть лишняя аномальная хромосома. Характеристики зависят от происхождения дополнительного генетического материала. Синдром кошачьего глаза и isodicentric хромосомы 15 синдром (или Idic15) оба вызваны нештатными маркера хромосомой, как синдром Паллистер-Киллиан .
- Синдром Triple-X (XXX). XXX девушки, как правило, высокие и худые и чаще страдают дислексией.
- Синдром Тернера (X вместо XX или XY). При синдроме Тернера женские половые признаки присутствуют, но недостаточно развиты. Женщины с синдромом Тернера часто имеют низкий рост, низкий рост волос, аномальные черты глаз и развитие костей, а также вид «вдавленной» груди.
- Синдром Вольфа – Хиршхорна , который вызван частичной делецией короткого плеча хромосомы 4. Он характеризуется задержкой роста, задержкой развития моторики, чертами лица «греческого шлема» и легкими или серьезными проблемами психического здоровья.
- XYY-синдром . Мальчики XYY обычно выше своих братьев и сестер. Подобно XXY мальчикам и XXX девочкам, они чаще испытывают трудности в обучении.
Анеуплоидия спермы
Воздействие на мужчин определенного образа жизни, окружающей среды и / или профессиональных опасностей может увеличить риск анеуплоидных сперматозоидов. В частности, риск анеуплоидии увеличивается при курении табака и воздействии бензола, инсектицидов и перфторированных соединений на рабочем месте. Повышенная анеуплоидия часто связана с повышенным повреждением ДНК в сперматозоидах.
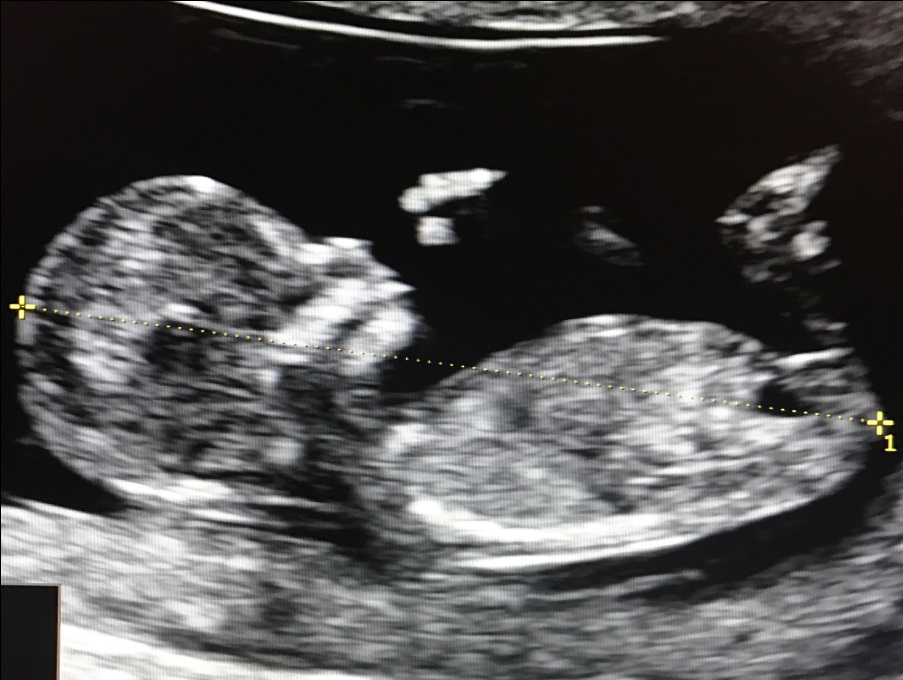
Почему рождаются дети Дауны: фото необычных малышей
Детки с синдромом Дауна не так уж и редко встречаются в нашей жизни. Совсем недавно их число было достаточно большим. Однако благодаря современной медицине даунята стали рождаться реже.
Данное заболевание возникает, если вместо сорока шести пар хромосом появляется сорок семь. Эта лишняя хромосома образуется в двадцать третьей паре. Поэтому синдром Дауна еще называют трисомией по двадцать первой паре хромосом.
В нормальном состоянии двадцать три мужские и двадцать три женские хромосомы соединяются, и складываются в сорок шесть хромосом. Однако может возникнуть мутация, когда один из родителей выделит клетку, в которой будет на одну хромосому больше. В этом случае появляется синдром Дауна.
Синдром Дауна может стать причиной случайной мутацией яйцеклетки или сперматозоида, аномальное деление клеток в процессе развития зародыша или плохая наследственность у родителей. В зависимости от этого выделяют три типа даунизма.
Типы даунизма:
- Простая трисомия – это самый распространенный тип синдрома Дауна. Он встречается в девяносто четырех процентов случаев даунизма. В данном случае болезнь происходит из-за случайной мутации родительских клеток. Трисомия наблюдается во всех клетках.
- Мозаичная форма даунизма самая редкая. В этом случае на начальной стадии формирования зародыша в одной из клеток появляется трисомия. В этом случае мутация появляется лишь в тех клетках, на которые разделилась с трисомией.
- Транслокационная форма даунизма стоит на втором месте по частоте появления. В этом случае 21 хромосома прикрепляется к одной из здоровых хромосом и начинает транслировать мутированные клетки. В этом случае причиной такой неприятности является плохая наследственность отца или матери.
Дети с синдромом Дауна рождаются достаточно часто и выглядят необычно. И причины подобной мутации пока не изучены. Ученые считают, что велика вероятность рождения таких детей у женщин после тридцати пяти лет, и у мужчин после сорока пяти.
Вам будет интересно узнать, что если женщина рождает дауненка, то его однояйцовый близнец тоже будет иметь этот синдром. При этом очень редко случается так, что с таким синдромом рождаются оба разнояйцовых близнеца. Вероятность, что если один ребенок в семье имеет такой синдром, то следующий ребенок родится с ним, ничтожно мала.
Прогноз для детей с синдромом Эдвардса
из различных независимых исследований50 – 55%сердцеполной трисомией 18половая зрелость обычно не наступаетдаже механической, не требующей особых навыковпри отсутствии пороков системы пищеваренияЧаще всего пороки развития наблюдаются в следующих органах и системах:
- опорно-двигательный аппарат (кости и суставы, включая череп);
- сердечно-сосудистая система;
- центральная нервная система;
- пищеварительная система;
- мочеполовая система;
- другие нарушения.
Сердечно-сосудистая система
сердечной недостаточностиНаиболее частыми аномалиями со стороны сердечно-сосудистой системы являются:
- незаращение межпредсердной перегородки;
- незаращение межжелудочковой перегородки;
- сращение створок клапанов (или, наоборот, их недоразвитие);
- коарктация (сужение) аорты.
пороки сердца
Пищеварительная система
Наиболее частыми пороками развития со стороны пищеварительной системы являются:
- дивертикул Меккеля (слепой отросток в тонкой кишке);
- атрезия пищевода (зарастание его просвета, из-за чего пища не проходит в желудок);
- атрезия желчных путей (накопление желчи в пузыре).
Типы половых хромосом
Существует множество форм, которые могут принимать половые хромосомы, и множество способов определения пола. Два основных способа, которыми гетероморфные половые хромосомы могут определять пол, известны как системы XY и ZW.
X и Y хромосомы
В системе XY мужчины содержат одну X и одну Y-хромосому, в то время как женщины содержат две X-хромосомы. Таким образом, мужчины считаются гетерогаметными – они могут производить два разных типа гамет в зависимости от того, несет ли сперма хромосому X или Y. Самки гомогаметны – все их яйца несут одну Х-хромосому. Многие приматы, включая людей, используют систему определения пола XY.
Вариант этого – метод, используемый некоторыми кузнечиками. Здесь мужчины имеют только одну Х-хромосому и не имеют Y-хромосому. В таких системах считается, что самец или самка развиваются на основе соотношения между Х-хромосомами и числом наборов аутосом, Например, если диплоидный индивид имеет две Х-хромосомы, он превращается в женщину, а самцы возникают из диплоидов, у которых есть одна Х-хромосома
Влияние соотношений особенно важно при определении пола у плодовых мух Drosophila melanogaster и круглых червей C. elegans, где особи XXY или XXYY – женщины, а XO – мужчины
Это противоречит случаю у людей, когда простое присутствие одной Y-хромосомы придает мужественность, независимо от количества Х-хромосом или соотношения между половыми хромосомами и аутосомами.
До недавнего времени было широко распространено мнение, что Y-хромосома у приматов подвергается быстрому ген потеря и то, что хромосома полностью исчезнет приблизительно через десять миллионов лет, приводя приматов к системе определения пола XX / XO. В настоящее время это оспаривается, и исследование эволюции половых хромосом ведет к новым открытиям.
W и Z хромосомы
Птицы, некоторые рыбы, рептилии и даже некоторые беспозвоночные подвергаются определению пола по методу ZW. Здесь мужчины гомогаметны (ZZ), а женщины несут две разные половые хромосомы (ZW). Иногда W-хромосома может полностью отсутствовать, например у некоторых видов бабочек, и ZO развиваются в самок. В других случаях присутствие W-хромосомы даже не обязательно для развития в самок.
Прокариоты
В прокариоты — бактерии и археи — как правило , имеют одну круговую хромосому , но существует множество вариаций. Хромосомы большинства бактерий, которые некоторые авторы предпочитают называть генофорами , могут иметь размер от 130 000 пар оснований в эндосимбиотических бактериях Candidatus Hodgkinia cicadicola и Candidatus Tremblaya princeps до более 14 000 000 пар оснований у обитающей в почве бактерии Sorangium cellulosum . Спирохеты из рода Borrelia являются заметным исключением из этого порядка, поскольку такие бактерии, как Borrelia burgdorferi , вызывающие болезнь Лайма , содержат одну линейную хромосому.
Структура в последовательностях
Прокариотические хромосомы имеют менее последовательную структуру, чем эукариотические. Бактерии обычно имеют одну точку ( источник репликации ), из которой начинается репликация, тогда как некоторые археи содержат несколько источников репликации. Гены прокариот часто организованы в опероны и обычно не содержат интронов , в отличие от эукариот.
Упаковка ДНК
Прокариоты не имеют ядер. Вместо этого их ДНК организована в структуру, называемую нуклеоидом . Нуклеоид представляет собой отдельную структуру и занимает определенную область бактериальной клетки. Эта структура, однако, динамична, поддерживается и модифицируется за счет действия ряда гистоноподобных белков, которые связаны с бактериальной хромосомой. У архей ДНК в хромосомах еще более организована: ДНК упакована в структуры, подобные эукариотическим нуклеосомам.
Некоторые бактерии также содержат плазмиды или другую внехромосомную ДНК . Это кольцевые структуры в цитоплазме, которые содержат клеточную ДНК и играют роль в горизонтальном переносе генов . У прокариот (см. Нуклеоиды ) и вирусов ДНК часто плотно упакована и организована; в случае архей — по гомологии с эукариотическими гистонами, а в случае бактерий — с гистоноподобными белками.
Бактериальные хромосомы имеют тенденцию быть привязанными к плазматической мембране бактерий. В приложениях молекулярной биологии это позволяет изолировать плазмидную ДНК центрифугированием лизированных бактерий и осаждением мембран (и прикрепленной ДНК).
Прокариотические хромосомы и плазмиды, как и эукариотическая ДНК, обычно сверхспиральны . ДНК сначала должна быть переведена в расслабленное состояние для доступа к транскрипции , регуляции и репликации .
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала по 21-й хромосоме, либо полностью (трисомия), либо частично (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от степени копии, генетической истории и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например был обнаружен у обезьян и мышей). Совсем недавно[когда?
] исследователи[кто? ] вывели трансгенных мышей с наличием 21-й человеческой хромосомы (в дополнение к стандартному набору мышей). Добавление генетического материала может проводиться в разных направлениях. Типичный человеческий кариотип обозначается как 46,XY (мужской) или 46,XX (женский) (различие в поле несёт Y-хромосома).
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме.
Синдром Дауна и сходные хромосомные аномалии чаще встречаются у детей, рождённых немолодыми женщинами. Точная причина этого неизвестна, но, по-видимому, она как-то связана с возрастом яйцеклеток матери.
Трисомия происходит из-за того, что во время мейоза хромосомы не расходятся. При слиянии с гаметой противоположного пола у эмбриона образуется 47 хромосом, а не 46, как без трисомии.
Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских.
Мозаицизм
Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является как правило более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики.
По данному типу синдром появляется в 1—2 % случаев.
Робертсоновские транслокации
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться за счёт робертсоновских транслокаций в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й ). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции, нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна
. Это не зависит от возраста матери и показывает скорее равную роль родительских организмов в появлении синдрома Дауна. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Дублирование части хромосомы 21
Очень редко, но части хромосом могут делиться. Это создаст дополнительные копии некоторых, но не всех генов из 21-й хромосомы. Если продублируются фрагменты, обусловливающие физические и психологические проявления синдрома Дауна, то ребёнок родится с этим синдромом. Такое дублирование происходит крайне редко и не существует оценки периодичности данного явления.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.
Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет.
Синдром Ди Джорджи
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
Синдром Ангельмана
Причины генетической патологии
ДНКнуклеотидовбелок, какой-либо фермент или рецептор организмаВсе хромосомы в организме человека делятся на два вида:
- Аутосомы. Аутосомы – это хромосомные пары с 1 по 22. Они несут большой объем генетической информации и могут быть различных размеров. При синдроме Дауна у больных наблюдается утроение аутосомы под номером 21.
- Половые хромосомы. Половые хромосомы обозначаются цифрами Х и Y. Они предопределяют пол человека (XX – девочка, XY – мальчик). Условно эти хромосомы объединяют в 23-ю пару, хотя Х и Y не похожи друг на друга ни размером, ни формой, ни набором генов.
в последовательности нуклеотидовдля женщиндля мужчиндве хромосомы, составляющие пару, соединяются не в виде буквы Х, а в виде буквы VВ зависимости от характера хромосомной мутации различают следующие виды болезни:
- Полная трисомия 21. Полная трисомия 21 предполагает, что у ребенка в каждой клетке организма имеется целая дополнительная хромосома. Таким образом, общее количество ее копий – 3. Частота данного варианта составляет 90 – 95%. Эта форма является наиболее тяжелой. У пациента наблюдается избыток всех генов, закодированных в этой молекуле ДНК. Как правило, нарушения внутриутробного развития у них встречаются чаще, а умственная отсталость более выражена. Полная трисомия возникает, если один из родителей передает ребенку не одну, а две хромосомы 21. Тогда при слиянии с третьей 21-й хромосомой (от второго родителя) возникает трисомия. Зигота (первая клетка, из которой возникает зародыш) уже содержит дефект. Дальнейшее ее деление объясняет, что все дочерние клетки будут похожи на нее.
- Мозаичная форма. При мозаичной форме механизм появления хромосомного дефекта несколько иной. Обе родительские гаметы (половые клетки) имели нормальное количество хромосом. После их слияния образовалась нормальная зигота с кариотипом 46, ХХ или 46, XY. В процессе деления этой первоначальной клетки ДНК распределилось неправильно. Часть клеток организма получилась с нормальным кариотипом, а часть – с кариотипом синдрома Дауна. Такая аномалия встречается довольно редко (3 – 5% случаев данного заболевания). Прогноз при ней лучше, так как здоровые клетки отчасти компенсируют генетический дефект. Ребенок все равно родится с синдромом Дауна и видимым отставанием в развитии. Однако выживаемость таких детей значительно выше. У них редко встречаются тяжелые пороки развития внутренних органов, несовместимые с жизнью.
- Семейный синдром Дауна. Семейный синдром Дауна – это весьма редкий генетический дефект (менее 2% случаев). При нем один из родителей имеет небольшие отклонения. Часть хромосомы 21 (а именно, критический участок) прикрепляется к другой хромосоме (обычно к 14-й). Таким образом, на 14 хромосоме содержится больше генетической информации, чем должно быть в норме. У человека при этом обычно нет видимых изменений (симптомов синдрома Дауна). Однако все половые гаметы, которые производит его организм, содержат этот дополнительный участок хромосомы 21. Очень высока вероятность, что в процессе образования зиготы такая гамета вызовет появление дополнительной 21-й хромосомы. Таким образом, дети у человека с подобным дефектом часто рождаются с синдромом Дауна. Из-за этой аномалии, передающейся потомству, данную форму болезни назвали семейной.
- Частичная трисомия 21. При частичной трисомии 21 у пациента обнаруживается не вся дополнительная хромосома, а лишь ее фрагмент с критическим участком. Из-за этого у ребенка развивается синдром Дауна в более легкой форме (однако все основные симптомы все равно присутствуют). Механизм такого дефекта чем-то похож на семейную форму болезни, но синдром не будет передаваться по наследству. Встречается этот вариант болезни очень редко.
На образование аномальных гамет могут повлиять следующие факторы:
- экологическая обстановка;
- некоторые медикаменты;
- курение;
- алкоголизм;
- радиация;
- некоторые заболевания половой сферы.
зачать ребенкаВероятность рождения ребенка в зависимости от возраста матери выглядит следующим образом:
- 0,064% для женщин, рожающих в возрасте 20 – 24 лет;
- 0,1% — для женщин в возрасте 25 – 30 лет;
- 0,17% — для женщин в возрасте 31 – 35 лет;
- 0,47% – для женщин 36 – 40 лет;
- 0,78% — для женщин 41 – 45 лет;
- до 5,25% — у женщин старше 45 лет (синдром Дауна – у каждого двадцатого ребенка).