Синдром дауна (монголизм)
Содержание:
- Типы хромосом. Принципы классификации хромосом. Денверская и Парижская классификации хромосом, их сущность.
- Синдром Ангельмана
- Симптомы[править | править код]
- Трисомии
- Хромосомные расстройства
- Общие вопросы
- Связь хромосомных аномалий и злокачественных образований
- Как выглядят новорожденные с синдромом Эдвардса?
- Лечение ЗПРР при хромосомных заболеваниях.
- Общая частота
- Зачем знать о рисках наследственных заболеваний
- Этиология хромосомных заболеваний
- Аниридия
- Цитогенетическая полоса
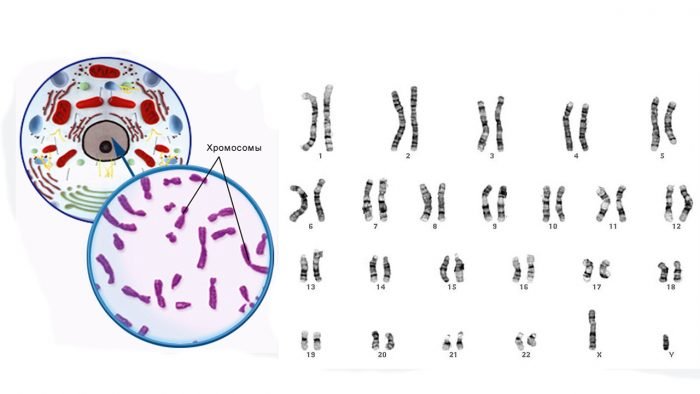
Типы хромосом. Принципы классификации хромосом. Денверская и Парижская классификации хромосом, их сущность.
Метафазная
хромосома состоит
из двух продольных нитей ДНП – хроматид,
соединенных друг с другом в области
первичной перетяжки (центромеры).
Центромера делит тело хромосомы на два
плеча. В зависимости от расположения
центромеры различают следующие типы
хромосом:
акроцентрические – центромера смещена к одному концу
хромосомы и одно плечо очень короткое;
субметацентрические –
центромера смещена от середины
хромосомы, и плечи имеют разную длину;
метацентрические –
центромера расположена посередине, и
плечи примерно одинаковой длины.
Участок
каждого плеча вблизи центромеры
называется – проксимальным, удаленный
от нее –дистальным. Концевые отделы
дистальных участков называются
теломерами. Теломеры препятствуют соединению концевых
участков хромосом. При потере этих
участков наблюдаются хромосомные
перестройки. Некоторые хромосомы могут
иметь вторичные
перетяжки ,
отделяющие от тел хромосомы участок,
называемый спутником .
Правила
хромосом.
Правило
постоянства числа хромосом.
Правило
парности хромосом.
Правило
индивидуальности хромосом.
Правило
непрерывности хромосом.
Денверская
классификация хромосом, которая
помимо размеров хромосом, учитывает их
форму, положение центромеры и наличие
вторичных перетяжек и спутников. 23 пары
хромосом человека разбили на 7 групп от
A
до G.
Важным параметром является центромерный
индекс (ЦИ), который отражает отношение
(в %) длины короткого плеча к длине всей
хромосомы.
К группе A относят 1-3 хромосомы. Это большие
метацентрические и субметацентрические
хромосомы, их центромерный индекс от
38-49.
Группа B (4 и 5 пары). Это большие субметацентрические
хромосомы, ЦИ 24-30.
Группа C (6-12 пары). Хромосомы среднего размера,
субметацентрические, ЦИ 27-35. К этой
группе относят и Х-хромосому.
Группа D (13-15 пары). Хромосомы акроцентрические,
сильно отличаются от всех других хромосом
человека, ЦИ около 15.
Группа E (16-18 пары). Относительно короткие,
метацентрические или субметацентрические,
ЦИ 26-40.
Группа F (19-20 пары): две короткие, субметацентрические
хромосомы, ЦИ 36-46.
Группа G (21-22 пары): это маленькие акроцентрические
хромосомы, ЦИ 13-33. К этой группе относят
и Y-хромосому.
В
основе Парижской
классификации хромосом человека (1971 г.) лежат методы специальной
дифференциальной их окраски, при которой
каждой хромосоме выявляется характерный
только для нее порядок чередования
поперечных светлых и темных сегментов.
Различные
типы сегментов обозначают по методам,
с помощью которых они выявляются наиболее
отчетливо (Q-сегменты, G-сегменты,
Т-сегменты, S-сегменты). Каждая хромосома
человека содержит свойственную только
ей последовательность полос, что
позволяет идентифицировать каждую
хромосому. Хромосомы спирализованы
максимально в метафазе, менее спирализованы
в профазе и прометафазе, что позволяет
выделить большее число сегментов, чем
в метафазе.
На
метафазной хромосоме (рис. 59) приводятся
символы, которыми принято обозначать
короткое и длинное плечо, а также
расположение районов и сегментов. В
настоящее время существуют ДНК-маркеры
или зонды, с помощью которых можно
определить изменение определенного,
даже очень маленького, сегмента в
хромосомах (цитогенетические карты).
На международном конгрессе генетики
человека в Париже в 1971 г. (Парижская
конференция по стандартизации и
номенклатуре хромосом человека) была
согласована система символов для более
краткого и однозначного обозначения
кариотипов. При
описании кариотипа :
•
указывается общее
число хромосом и набор половых хромосом ,
между ними ставится запятая (46, XX; 46, XY);
• отмечается какая хромосома лишняя или какой не
хватает (это указывается ее номером 5, 6 и др., или
буквами данной группы А, В и др.); знаком
«+» указывают на увеличение количества
хромосом , знаком
«-» указывают на отсутствие
данной хромосомы 47, XY,+ 21;
•
плечо хромосомы, в котором произошло
изменение ( удлинение
короткого плеча указывается символом ( р+); укорочение
(р-); удлинение
длинного плеча указывается символом
(q+); укорочение (q-); •
символы перестроек ( транслокация
обозначается t, а делеция — del )
помещают перед номерами вовлеченных
хромосом, а перестроечные хромосомы
заключают в скобки. Наличие двух
структурно-аномальных хромосом
обозначается точкой с запятой (;) или
нормальной дробью (15/21).
Синдром Ангельмана
Симптомы[править | править код]
Симптомы 49, XXXXY схожи с симптомами синдрома Клайнфелтера и 48, XXXY, однако они обычно намного более выражены при синдроме 49, XXXXY. Анеуплоидия часто приводит к летальному исходу, но в этом случае существует «инактивация Х», при которой эффект дополнительных Х хромосом значительно снижается.
Репродуктивное
Лица с синдромом 49, XXXXY, как правило, демонстрируют неразвитые вторичные половые признаки и стерильность.
Гипопластические гениталии.
Физические
Мужчины с таким кариотипом как правило, имеют многочисленные скелетные аномалии. Эти скелетные аномалии включают в себя:
- Genu Valgum
- Pes Cavus
Клинодактилия
Также:
Психические
Как и при синдроме Дауна, психические проявления синдрома 49, XXXXY различны. Типичными являются нарушения речи и неадекватные поведенческие проблемы. В одном исследовании рассматривались мужчины с диагнозом 48, XXYY, 48, XXXY и 49, XXXXY. Они обнаружили, что мужчины с 48, XXXY и 49, XXXXY функционируют на гораздо более низком когнитивном уровне, чем мужчины их возраста. Эти мужчины также имеют тенденцию проявлять более незрелое поведение для своего возраста; повышенные агрессивные тенденции были также процитированы в этом исследовании.
Патофизиология
Как видно из названия, у человека с этим синдромом есть одна Y-хромосома и четыре X-хромосомы на 23-й паре, таким образом, имеется сорок девять хромосом, а не нормальные сорок шесть. Как и в большинстве категорий анеуплоидных расстройств, синдром 49, XXXXY часто сопровождается умственной отсталостью. Его можно рассматривать как форму или вариант синдрома Клайнфелтера (47, XXY). Люди с этим синдромом, как правило, мозаичные, 49, XXXXY / 48, XXXY.
Это генетическое, но не наследственное заболевание, что означает, что, хотя гены родителей вызывают синдром, существует небольшая вероятность того, что синдром у более чем одного ребенка. Вероятность наследования заболевания составляет около одного процента.
Трисомии
В материале выкидышей трисомии представляют более половины всех количественных хромосомных аберраций
Обращает на себя внимание то, что в случаях моносомий недостающей хромосомой обычно оказывается X-хромосома, а в случаях избыточных хромосом, дополнительная хромосома чаще всего оказывается аутосомой
Точная идентификация дополнительной хромосомы стала возможна благодаря методу G-бэндинга. Исследования показали, что все аутосомы могут принимать участие в нон-дисджанкшн (см. таблицу)
Обращает на себя внимание, что три хромосомы, чаще всего встречающиеся при трисомиях новорожденных (15-я, 18-я и 21-я) чаще всего обнаруживаются и при летальных трисомиях у зародышей. Вариации относительных частот различных трисомий у зародышей отражают во многом сроки, на которых происходит гибель зародышей, поскольку, чем более летальной является комбинация хромосом, тем на более ранних сроках происходит остановка развития, тем реже будет обнаруживаться такая аберрация в материалах выкидышей (чем меньше срок остановки развития, тем труднее обнаружить такой зародыш)
| Лишняя хромосома при летальных трисомиях у зародыша (данные 7 исследований: Буэ (Франция), Карр (Канада), Кризи (Великобритания), Дилл (Канада), Кадзи (Швейцария), Такахара (Япония), Теркелсен (Дания)) | ||
|---|---|---|
| Дополнительная аутосома | Количество наблюдений | |
| A | 1 | |
| 2 | 15 | |
| 3 | 5 | |
| B | 4 | 7 |
| 5 | ||
| C | 6 | 1 |
| 7 | 19 | |
| 8 | 17 | |
| 9 | 15 | |
| 10 | 11 | |
| 11 | 1 | |
| 12 | 3 | |
| D | 13 | 15 |
| 14 | 36 | |
| 15 | 35 | |
| E | 16 | 128 |
| 17 | 1 | |
| 18 | 24 | |
| F | 19 | 1 |
| 20 | 5 | |
| G | 21 | 38 |
| 22 | 47 |
Хромосомные расстройства
Следующие состояния вызваны изменениями структуры или количества копий хромосомы 7:
Синдром Вильямса вызывается делецией генетического материала из части длинного (q) плеча хромосомы 7. Удаленная область, которая расположена в позиции 11.23 (записывается как 7q11.23), обозначается как критическая область синдрома Вильямса. . Этот регион включает более 20 генов, и исследователи полагают, что характерные черты синдрома Вильямса, вероятно, связаны с потерей нескольких генов в этом регионе.
Хотя несколько конкретных генов, связанных с синдромом Вильямса, были идентифицированы, связь между большинством генов в удаленной области и признаками и симптомами синдрома Вильямса неизвестна.
Другие изменения числа или структуры хромосомы 7 могут вызывать задержку роста и развития, психические расстройства, характерные черты лица, аномалии скелета, задержку речи и другие проблемы со здоровьем. Эти изменения включают дополнительную копию части хромосомы 7 в каждой клетке (частичная трисомия 7) или недостающий сегмент хромосомы в каждой клетке (частичная моносомия 7). В некоторых случаях несколько строительных блоков ДНК ( нуклеотидов ) удаляются или дублируются в части хромосомы 7. Также возможна кольцевая структура, называемая кольцевой хромосомой 7. Кольцевая хромосома возникает, когда оба конца сломанной хромосомы воссоединяются.
Общие вопросы
Связь хромосомных аномалий и злокачественных образований
Как правило, исследование клеток злокачественных новообразований приводит к обнаружению видимых в микроскоп хромосомных аномалий. Сходные результаты дает проверка при лейкозе, лимфоме и ряде других заболеваний.
В частности, для лимфом нередким случаем является обнаружение транслокации, сопровождающейся разрывом внутри или рядом с локусом тяжелой цепи иммуноглобулина (14 хромосома). При этом ген MYC перемещается с 8 хромосомы на 14.
Для миелолейкоза в большинстве случаев (свыше 95%) фиксируется транслокация между 22 и 9 хромосомами, вызывающая появление характерной «филадельфийской» хромосомы.
Бластный криз в процессе развития сопровождается появлением в кариотипе последовательных хромосомных аномалий.
Методами дифференциального окрашивания с последующим наблюдением в микроскоп, а также при помощи молекулярно-генетических способов тестирования, можно своевременно выявлять хромосомные аномалии при различных лейкозах. Эта информация помогает сделать прогноз развития, по ней уточняется диагноз и корректируется терапия.
Для распространенных солидных опухолей, таких, как рак толстой кишки, рак молочной железы и т.д. обычные цитогенетические методы применимы с некоторыми ограничениям. Тем не менее, характерные для них хромосомные аномалии также были выявлены. Имеющиеся в опухолях отклонения часто связаны с генами, отвечающими за процесс нормального роста клеток. Из-за амплификации (образования множественных копий) гена иногда отмечается формирование мелких мини-хромосом в клетках новообразований.
В некоторых случаях появление злокачественного образования вызывает потеря гена, который должен обеспечивать подавление пролиферации. Причин может быть несколько: делеции и разрыв в процессе транслокации являются наиболее частыми. Мутации такого рода принято считать рецессивными, поскольку наличие даже одной нормальной аллели обычно обеспечивает достаточный контроль роста. Нарушения могут появляться или наследоваться. Если же в геноме отсутствует нормальная копия гена, то пролиферация перестает зависеть от регулирующих факторов.
Таким образом, наиболее значимыми хромосомными аномалиями, влияющими на возникновение и рост злокачественных новообразований, являются следующие типы:
— транслокации, поскольку они могут привести к нарушению нормального функционирования генов, отвечающих за пролиферацию (либо вызвать их усиленную работу);
— делеции, которые наряду с прочими рецессивными мутациями вызывают изменения в процессе регуляции роста клетки;
— рецессивные мутации, из-за рекомбинации становящиеся гомозиготными и оттого проявляющиеся в полной мере;
— амплификации, стимулирующие пролиферацию клеток опухоли.
Выявление указанных мутаций в ходе генетической диагностики может указывать на повышенный риск развития злокачественных новообразований.
Как выглядят новорожденные с синдромом Эдвардса?
Новорожденные с синдромом Эдвардса имеют следующие характерные аномалии развития:
- изменение формы черепа;
- изменение формы ушных раковин;
- аномалии развития неба;
- стопа-качалка;
- аномальная длина пальцев;
- изменение формы нижней челюсти;
- сращение пальцев;
- аномалии развития половых органов;
- флексорное положение кистей;
- дерматоглифические признаки.
Аномалии развития неба
его можно прощупать посередине твердого неба языкомСуществует несколько вариантов данного дефекта:
- незаращение мягкого неба (задняя, глубокая часть неба, которая нависает над глоткой);
- частичное незаращение твердого неба (щель не тянется на протяжении всей верхней челюсти);
- полное незаращение твердого и мягкого неба;
- полное незаращение неба и губы.
даже при закрытом ртепочти 20% новорожденныхдо 65% новорожденных
Изменение формы нижней челюсти
микрогенияУ новорожденных с микрогнатией обычно быстро появляются следующие проблемы:
- невозможность долго держать рот закрытым (подтекание слюны);
- затруднения при кормлении;
- позднее развитие зубов и неправильное их расположение.
Дерматоглифические признаки
аномальные узоры и складки на коже ладонейОсновными дерматоглифическими признаками синдрома Эдвардса являются:
- дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
- кожная складка между последней (ногтевой) и предпоследней (срединной) фалангами пальцев отсутствует;
- у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан).
Лечение ЗПРР при хромосомных заболеваниях.
Основой лечения является уникальная методика патогенетической терапии речевых расстройств при хромосомной патологии — биофизическая активация нейромоторных структур, основу которого составляет щадящая стимуляция проводников нервной системы микротоками с использованием нейрофизиологического прибора. Метод лечения базируется как на активации самих речевых центров, так и на восстановлении нарушенных связей между центрами и полушариями головного мозга. Помимо этого, восстанавливаются разрозненные связи речевых центров с другими областями мозга, участвующими в реализации речевой функции. В процессе лечения формируется физиологичное, последовательное взаимодействие всех зон мозга, связанных с речепродукцией. В результате появляется речь.
Проведение биофизической активации сочетается с дополнительными методиками лечения, такими как — лимфомежклеточная терапия, которая применяется для регулирования интегративной деятельности и восполнения дефицита энергетической системы мозга и позволяющая применять малые дозы церебропротекторов, которые вводятся эндолимфатически и попадают в ткани головного мозга, минуя гематоэнцефалический барьер.
В качестве другого способа использования препаратов с нейротрофическим и антиоксидантным действием применяется методика эндоназального электрофореза кортексина, что позволяет вводить лекарственные препараты непосредственно в ткани головного мозга.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Также для успешного лечения различных форм ЗПРР специалистами применяется одно из достижений современной науки — метод аудиовокальной терапии RUSTOMATIS. Прибор использует звукозаписи высокочастотных и низкочастотных компонентов. При чередовании такой музыки путем напряжения и расслабления у ребенка тренируется аппарат среднего уха – молоточек и стремечко, с помощью чего расширяется диапазон восприятия внешних факторов, увеличивается концентрация внимания, в мозг поступает новая информация и, как следствие исчезают многие нарушения и расстройства.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Общая частота
При оценке результатов больших серий анализов следует иметь в виду следующее. На результаты исследований подобного рода могут оказать значительное влияние следующие факторы: способ сбора материала, относительная частота более ранних и более поздних выкидышей, доля материала искусственных абортов в исследовании, часто не поддающаяся точной оценке, успех культивирования клеточных культур абортуса и хромосомного анализа материала, тонкие методы обработки мацерированного материала.
Общая оценка частоты хромосомных аберраций при невынашивании беременности составляет около 60%, а в первом триместре беременности — от 80 до 90%. Как будет показано ниже, анализ, основанный на стадийности развития зародыша, позволяет сделать гораздо более точные выводы.
Зачем знать о рисках наследственных заболеваний
Человеческий организм состоит из триллионов клеток. Каждая играет определенную роль: отвечает за производство ферментов для расщепления пищи, транспортировку кислорода в крови или выполняет другие необходимые для жизни функции.
Большинство клеток человека содержит 23 хромосомы от матери и 23 — от отца. Всего 46 хромосом
Гены — участки ДНК, которые содержатся в хромосомах и передаются по наследству. У человека по разным оценкам 20–25 тысяч генов. Они изменяются при передаче потомству — во время деления клетки получают по одной хромосоме от каждой пары.
Изменения в генах, которые приводят к нарушению работы важных белков и развитию заболеваний, называются патогенными вариантами генов. В зависимости от степени изменений в генах заболевания делят на следующие категории:
| Категория | Причина развития | Примеры заболеваний |
|---|---|---|
| Хромосомные заболевания | Вся хромосома или ее большие сегменты отсутствуют, дублируются или изменены. | Синдром Дауна, трисомия 13, трисомия 18, синдром Клайнфельтера и синдром Тернера |
| Моногенные заболевания | В гене происходит изменение, из-за которого он перестает работать | Муковисцидоз, серповидноклеточная анемия, синдром ломкой Х-хромосомы, мышечная дистрофия или болезнь Хантингтона |
| Многофакторные расстройства | Мутации нескольких генов. Часто в сочетании с факторами окружающей среды | Диабет, болезнь Альцгеймера, Паркинсона, ожирение |
| Митохондриальные нарушения | Редкие нарушения, вызванные вариантами гена в митохондриальной ДНК. Эти нарушения могут поражать любую часть тела, в том числе мозг и мышцы. | Болезнь Альцгеймера, мышечная дистрофия, болезнь Лу Герига, диабет и рак |
Наличие патогенного варианта гена только у матери или отца, не означает, что у ребенка обязательно проявится заболевание. Человек может быть здоровым носителем. Но если у обоих родителей одинаковый патогенный вариант гена, то риск рождения ребенка с заболеванием значительно повышается.
Одно из самых распространенных наследственных заболеваний в европейской популяции — муковисцидоз, при котором в легких и других органах скапливается слизь. Она блокирует легкие, поджелудочную железу и другие органы. По статистике в Европе один из 2 000–3 000 новорожденных появляется на свет с муковисцидозом. Только половина пациентов с этим диагнозом доживает до 18 лет, и только 5% — до 40 лет.
Наследственные заболевания часто не совместимы с жизнью, поэтому распространено мнение о том, что они не так часто встречаются. Но по статистике 1 из 50 людей рождается с моногенным заболеванием, в то время как у 1 из 263 есть хромосомные нарушения.
В мире живет более 70 000 людей с муковисцидозом.
При наличии у обоих родителей варианта гена, связанного с муковисцидозом, риск рождения ребенка с этим заболеванием — 25%. Если узнать об этом заранее с помощью генетического теста, можно вовремя принять необходимые меры, например, воспользоваться вспомогательными репродуктивными технологиями.
Этиология хромосомных заболеваний
К этиологическим факторам хромосомных патологий относятся все разновидности хромосомных мутаций. Кроме того, некоторые геномные мутации также способны оказывать подобное действие.
У человека встречаются делеции, дупликации, транслокации и инверсии, то есть все типы мутаций. При делеции и дупликации генетическая информация оказывается в недостаточном и избыточном количестве соответственно. Поскольку современными методами можно выявить отсутствие даже небольшой части генетического материала (на уровне гена), то провести четкую границу между генными и хромосомными заболеваниями практически невозможно.
Транслокации представляют собой обмен генетическим материалом, который происходит между отдельными хромосомами. Иными словами, происходит перемещение участка генетической последовательности на негомологичную хромосому. Среди транслокаций выделяют две важные группы – реципрокные и Робертсоновские.
Транслокации реципрокного характера без потери задействованных участков называются сбалансированными. Они, как и инверсии, не вызывают потери генной информации, поэтому не приводят к паталогическим эффектам. Тем не менее, при дальнейшем участии таких хромосом в процессе кроссинговера и редукции могут образовываться гаметы с несбалансированными наборами, обладающие недостаточным набором генов. Их участие в процессе оплодотворения приводит к тому, что у потомства развиваются те или иные наследственные синдромы.
Для Робертсоновских транслокаций характерно участие двух акроцентрических хромосом. В ходе процесса короткие плечи утрачиваются, а длинные сохраняются. Из 2 исходных хромосом формируется одна цельная, метацентрическая. Несмотря на потерю части генетического материала развития патологий в таком случае обычно не происходит, поскольку функции утраченных участков компенсируются аналогичными генами в остальных 8 акроцентрических хромосомах.
При концевых делециях (то есть при их утрате) может сформироваться кольцевая хромосома. У ее носителя, получившего такой генный материал от одного из родителей, отмечают частичную моносомию по концевым участкам. При разрыве через центромеру может сформироваться изохромосома, имеющая одинаковые по набору генов плечи (у обычной хромосомы они отличаются).
В некоторых случаях может развиваться однородительская дисомия. Она возникает, если при нерасхождении хромосом и оплодотворении возникнет трисомия, а после этого одна из трех хромосом будет удалена. Механизм этого явления в настоящее время не изучен. Однако в результате в хромосомном наборе появится две копии хромосомы одного родителя, в то время как часть генной информации от второго родителя будет утеряна.
Многообразие вариантов искажения хромосомного набора обуславливает различные формы заболеваний.
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Цитогенетическая полоса
Идеограммы G-бэндинга хромосомы 15 человека
Идеограмма G-бэндинга 15 хромосомы человека в разрешении 850 ударов в час. Длина полосы на этой диаграмме пропорциональна длине пары оснований. Этот тип идеограммы обычно используется в браузерах генома (например, Ensembl , UCSC Genome Browser ).
Шаблоны G-полос 15 хромосомы человека в трех различных разрешениях (400, 550 и 850). Длина полосы на этой диаграмме основана на идеограммах из ISCN (2013). Этот тип идеограммы представляет собой действительную относительную длину полосы, наблюдаемую под микроскопом в различные моменты митотического процесса .
| Chr. | Рука | Группа | ISCN начало |
Остановка ISCN |
пар оснований старта |
Остановка базовой пары |
Пятно | Плотность |
|---|---|---|---|---|---|---|---|---|
| 15 | п | 13 | 270 | 1 | 4 200 000 | гвар | ||
| 15 | п | 12 | 270 | 631 | 4 200 001 | 9 700 000 | стебель | |
| 15 | п | 11.2 | 631 | 1142 | 9 700 001 | 17 500 000 | гвар | |
| 15 | п | 11.1 | 1142 | 1382 | 17 500 001 | 19 000 000 | Acen | |
| 15 | q | 11.1 | 1382 | 1487 | 19 000 001 | 20 500 000 | Acen | |
| 15 | q | 11.2 | 1487 | 1773 | 20 500 001 | 25 500 000 | гнег | |
| 15 | q | 12 | 1773 | 1968 г. | 25 500 001 | 27 800 000 | gpos | 50 |
| 15 | q | 13,1 | 1968 г. | 2164 | 27 800 001 | 30 000 000 | гнег | |
| 15 | q | 13,2 | 2164 | 2284 | 30 000 001 | 30 900 000 | gpos | 50 |
| 15 | q | 13,3 | 2284 | 2524 | 30 900 001 | 33 400 000 | гнег | |
| 15 | q | 14 | 2524 | 2765 | 33 400 001 | 39 800 000 | gpos | 75 |
| 15 | q | 15.1 | 2765 | 2975 | 39 800 001 | 42 500 000 | гнег | |
| 15 | q | 15,2 | 2975 | 3065 | 42 500 001 | 43 300 000 | gpos | 25 |
| 15 | q | 15.3 | 3065 | 3245 | 43 300 001 | 44 500 000 | гнег | |
| 15 | q | 21,1 | 3245 | 3471 | 44 500 001 | 49 200 000 | gpos | 75 |
| 15 | q | 21,2 | 3471 | 3621 | 49 200 001 | 52 600 000 | гнег | |
| 15 | q | 21,3 | 3621 | 3846 | 52 600 001 | 58 800 000 | gpos | 75 |
| 15 | q | 22,1 | 3846 | 3982 | 58 800 001 | 59 000 000 | гнег | |
| 15 | q | 22,2 | 3982 | 4087 | 59 000 001 | 63 400 000 | gpos | 25 |
| 15 | q | 22.31 | 4087 | 4252 | 63 400 001 | 66 900 000 | гнег | |
| 15 | q | 22,32 | 4252 | 4357 | 66 900 001 | 67 000 000 | gpos | 25 |
| 15 | q | 22,33 | 4357 | 4507 | 67 000 001 | 67 200 000 | гнег | |
| 15 | q | 23 | 4507 | 4613 | 67 200 001 | 72 400 000 | gpos | 25 |
| 15 | q | 24,1 | 4613 | 4748 | 72 400 001 | 74 900 000 | гнег | |
| 15 | q | 24,2 | 4748 | 4808 | 74 900 001 | 76 300 000 | gpos | 25 |
| 15 | q | 24,3 | 4808 | 4928 | 76 300 001 | 78 000 000 | гнег | |
| 15 | q | 25,1 | 4928 | 5048 | 78 000 001 | 81 400 000 | gpos | 50 |
| 15 | q | 25,2 | 5048 | 5169 | 81 400 001 | 84 700 000 | гнег | |
| 15 | q | 25,3 | 5169 | 5379 | 84 700 001 | 88 500 000 | gpos | 50 |
| 15 | q | 26,1 | 5379 | 5649 | 88 500 001 | 93 800 000 | гнег | |
| 15 | q | 26,2 | 5649 | 5860 | 93 800 001 | 98 000 000 | gpos | 50 |
| 15 | q | 26,3 | 5860 | 6070 | 98 000 001 | 101 991 189 | гнег |