Пренатальная (дородовая) диагностика врожденной патологии плода
Содержание:
- Виды
- Что оценивается при УЗИ в 1 триместре
- Патогенез
- Фенотипические проявления некоторых летальных хромосомных аберраций
- Эпидемиологические и демографические данные
- Двойные аберрации
- Причины
- Показания для тестирования на ломкую Х-хромосому
- Возникновение
- Скрининг на генетические заболевания
- Возможные осложнения при родах
- Прогноз
- Причины и признаки
- Что после рождения?
- Симптомы
- Лечение
Виды
Медицинское современное сообщество делит синдром Эдвардса на следующие виды:
1) Разновидность полной трисомии — когда клетки пациента хранят внутри своего состава лишние хромосомы, считается одной из самых тяжелых и распространенных форм заболеваний;
2) Частичной трисомии – когда дополнительная хромосома образуется в патогенных клетках;
3) Мозаичной формы – когда чуть меньшее количество клеток пациентов обладают нестандартными характеристиками их характеристик, локализируемых в разнообразных областях внутреннего организма или частей тела, так и только в одном месте.
Что оценивается при УЗИ в 1 триместре
1. Копчико-теменной размер (КТР) плода
Этот показатель точно определяет срок гестации (беременности), особенно в случае, если женщина не помнит 1-й день последней менструации, либо если менструальный цикл у нее не регулярный. В заключении срок беременности выставляется по КТР плода, а не по дате последней менструации.
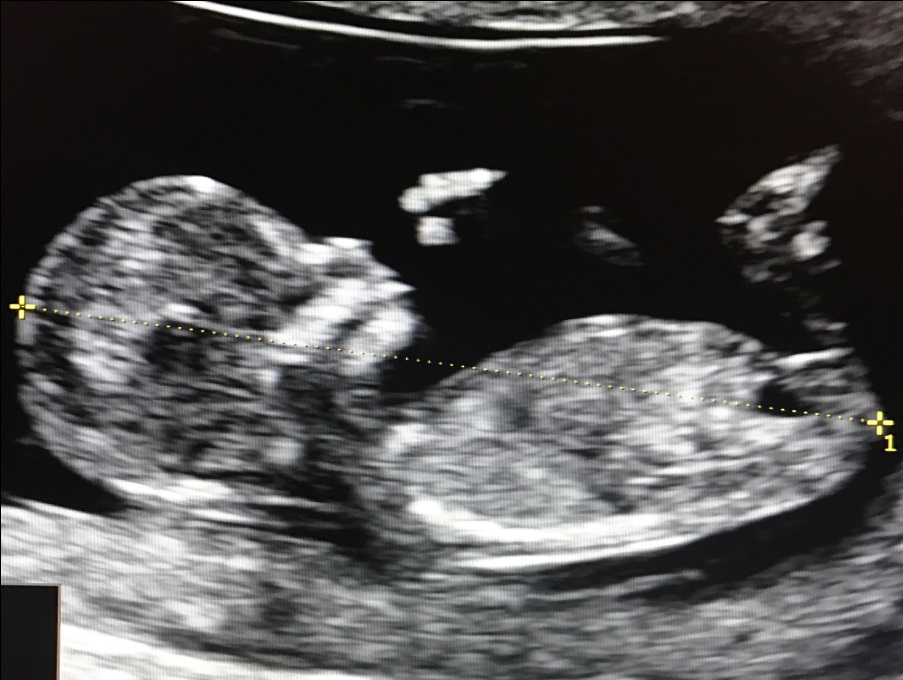
Правильное измерение КТР плода
2. Маркеры хромосомной патологии:
— толщина воротникового пространства (ТВП) – является основным признаком хромосомной патологии у плода. Патологической величиной считается увеличение ТВП больше 95-й процентили для каждого срока гестации. Каждое увеличение ТВП повышает риск существования хромосомной аномалии у плода.
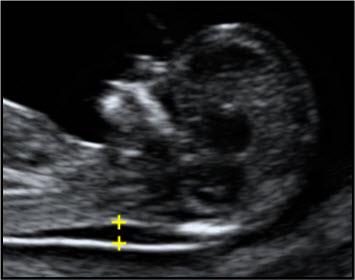
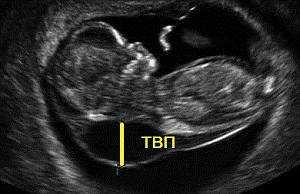
ТВП в норме ТВР при патологии
Важно понимать, что увеличение ТВП — это признак (маркер), но не точная диагностика хромосомных аномалий у плода. Определить наличие синдрома Дауна и других заболеваний у будущего ребенка позволяет только инвазивная диагностика с последующим генетическим анализом.. — носовая кость
У плодов с синдромом Дауна носовая кость может отсутствовать, либо быть уменьшенной (гипоплазированной). Очень редко такое может встречаться и у совершенно здоровых детей. Точный диагноз устанавливается только при помощи генетического анализа.
— носовая кость. У плодов с синдромом Дауна носовая кость может отсутствовать, либо быть уменьшенной (гипоплазированной). Очень редко такое может встречаться и у совершенно здоровых детей. Точный диагноз устанавливается только при помощи генетического анализа.
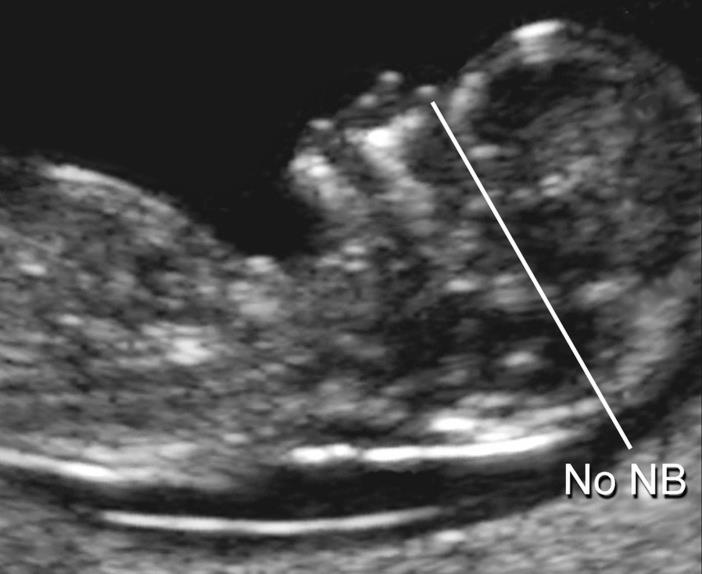
Нормальная носовая кость Отсутствие носовой кости
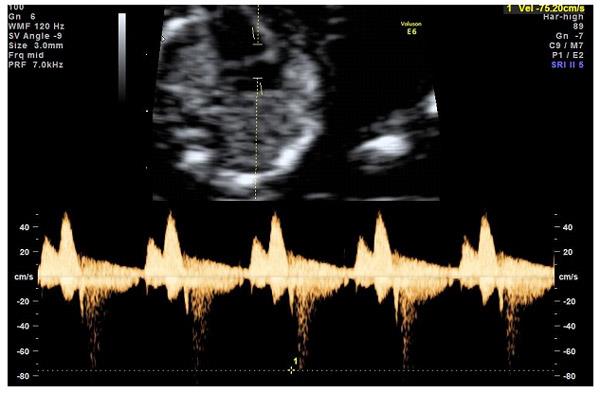
— кровоток в венозном протоке – это маленький сосуд в печени плода. При обратном (ретроградном) токе крови в данном сосуде можно предположить, что у плода хромосомный синдром, либо врожденный порок сердца.

Нормальный кровоток в венозном протоке
Но важно правильно получить этот кровоток и дать ему оценку. Для этого требуются определенные навыки и квалификация врача, которые подтверждаются ежегодной сертификацией FMF.. — кровоток через трикуспидальный клапан в сердце плода
Здесь ретроградный (обратный) кровоток тоже указывает на хромосомную патологию, либо может проявляться при врожденных пороках сердца.
— кровоток через трикуспидальный клапан в сердце плода. Здесь ретроградный (обратный) кровоток тоже указывает на хромосомную патологию, либо может проявляться при врожденных пороках сердца.
Ручка плода Мозг плода в виде “бабочки” в норме
Патогенез
Трисомия 18 – это генетическая патология. В каждой человеческой клетке содержится 46 хромосом – это нормальные показатели. Во время оплодотворения по 23 хромосомы женских и мужских половых клеток соединяются и дают в сумме именно такое количество хромосом. Иногда по неизвестным причинам происходят генетические мутации, и в результате появляется лишняя 47 хромосома, дополнительная в 18 паре. Лишняя хромосома у гамет появляется вследствие нерасхождения хромосом при мейотическом делении. В большинстве случаев – в 95% — именно лишняя хромосома становится причиной развития болезни Эдвардса. Но иногда – примерно в 2% случаев – сумма хромосом остается нормальной, но при этом 18 хромосома аномально удлиняется, что приводит к развитию врожденной патологии. Еще в 3% случаев отмечается мозаичная трисомия, при которой дополнительная хромосома присутствует не во всех клетках организма, а только в какой-то его части. Все три формы синдрома протекают по одному типу. Но все же более тяжелое течение отмечается при первой форме болезни.
Фенотипические проявления некоторых летальных хромосомных аберраций
Моносомии X обычно останавливаются в развитии к 6 неделям после зачатия. В двух третях случаев плодный пузырь размером 5—8 см не содержит зародыша, но существует шнурообразное образование с элементами эмбриональной ткани, остатками желточного мешка, плацента содержит субамниотические тромбы. В одной трети случаев плацента имеет такие же изменения, но обнаруживается морфологически неизмененный зародыш, погибший в возрасте 40—45 дней после зачатия.
При тетраплоидиях развитие останавливается к сроку 2-3 недели после зачатия, морфологически эта аномалия характеризуется «пустым плодным мешком».
При трисомиях наблюдаются различные типы аномалий развития, в зависимости от того, какая хромосома является лишней. Однако в подавляющем большинстве случаев развитие останавливается на очень ранних сроках, элементов зародыша не обнаруживается. Это классический случай «пустого плодного яйца» (анэмбрионии).
Трисомия 16, очень частая аномалия, характеризуется наличием маленького плодного яйца диаметром около 2,5 см, в полости хориона находится небольшой амниотический пузырек около 5 мм в диаметре и эмбриональный зачаток размером 1—2 мм. Чаще всего развитие останавливается на стадии эмбрионального диска.
При некоторых трисомиях, например, при трисомиях 13 и 14, возможно развитие зародыша до срока около 6 недель. Зародыши характеризуются циклоцефалической формой головы с дефектами закрытия верхнечелюстных холмиков. Плаценты гипопластичны.
Зародыши с трисомиями 21 (синдром Дауна у новорожденных) не всегда имеют аномалии развития, а если и имеют, то незначительные, не могущие служить причиной их гибели. Плаценты в таких случаев бывают бедны клетками, и представляются остановившимися в развитии на ранней стадии. Гибель зародыша в таких случаях представляется следствием плацентарной недостаточности.
Заносы. Сравнительный анализ цитогенетических и морфологических данных позволяет выделить два типа заносов: классический пузырный занос и эмбриональный триплоидный занос.
Выкидыши при триплоидиях имеют четкую морфологическую картину. Это выражается в сочетании полной или (чаще) частичной пузырной дегенерации плаценты и амниотического пузырька с зародышем, размеры которого (зародыша) очень малы по сравнению с относительно большим амниотическим пузырьком. Гистологическое исследование показывает не гипертрофию, а гипотрофию пузырно измененного трофобласта, образующего микрокисты в результате многочисленный инвагинаций.
Напротив, классический пузырный занос не затрагивает ни амниотический мешок, ни зародыш. В пузырьках обнаруживается избыточное образование синцитиотрофобласта с выраженной васкуляризацией. Цитогенетически большинство классических пузырных заносов имеет кариотип 46,XX. Проведенные исследования позволили установить хромосомные сбои, участвующие в образовании пузырного заноса. Было показано, что 2 X-хромосомы в классическом пузырном заносе идентичны и имеют отцовское происхождение. Наиболее вероятным механизмом развития пузырного заноса является истинный андрогенез, возникающий вследствие оплодотворения яйцеклетки диплоидным сперматозоидом, возникшим в результате сбоя второго мейотического деления и последующим полным выключением хромосомного материала яйцеклетки. С точки зрения патогенеза, такие хромосомные нарушения близки к нарушениям при триплоидии.
Эпидемиологические и демографические данные
Нечеткая клиническая симптоматика ранних самопроизвольных выкидышей приводит к тому, что достаточно большой процент выкидышей на малых сроках проходит незамеченным женщинами.
В случае клинически подтвержденных беременностей около 15% всех беременностей заканчивается выкидышем. Большая часть самопроизвольных выкидышей (около 80%) происходит в первом триместре беременности
Однако если принять во внимание тот факт, что выкидыши часто случаются спустя 4-6 недель после остановки развития беременности, можно сказать, что с первым триместром связано более 90% всех самопроизвольных выкидышей
Специальные демографические исследования позволили уточнить частоту внутриутробной смертности. Так, Френч и Бирман в 1953 — 1956 гг. регистрировали все беременности у женщин острова Канаи и показали, что из 1000 беременностей, диагностированных при сроке после 5 недель, 237 не увенчались рождением жизнеспособного ребенка.
Анализ результатов нескольких исследований позволил Леридону составить таблицу внутриутробной смертности, включающей в себя и неудачи оплодотворения (половой акт в оптимальные сроки — в течение суток после овуляции).
| Полная таблица внутри утробной смертности (на 1000 яйцеклеток, подвергшихся риску оплодотворения) (по Leridon, 1973) | ||
|---|---|---|
| Недели после зачатия | Остановка развития с последующим изгнанием | Процент продолжающихся беременностей |
| 16* | 100 | |
| 15 | 84 | |
| 1 | 27 | 69 |
| 2 | 5,0 | 42 |
| 6 | 2,9 | 37 |
| 10 | 1,7 | 34,1 |
| 14 | 0,5 | 32,4 |
| 18 | 0,3 | 31,9 |
| 22 | 0,1 | 31,6 |
| 26 | 0,1 | 31,5 |
| 30 | 0,1 | 31,4 |
| 34 | 0,1 | 31,3 |
| 38 | 0,2 | 31,2 |
| * — неудачи зачатия |
Все эти данные указывают на огромную частоту самопроизвольных выкидышей и на важную роль нарушений развития плодного яйца в этой патологии.
Эти данные отражают общую частоту нарушений развития, не выделяя среди них конкретные экзо- и эндогенные факторы (иммунологические, инфекционные, физические, химические и т. д.).
Важно отметить, что независимо от причины повреждающего воздействия, при исследовании материала выкидышей обнаруживается очень большая частота генетический нарушений (хромосомных аберраций (на сегодня изучены лучше всего) и генных мутаций) и аномалий развития, как, например, дефекты развития нервной трубки
Двойные аберрации
Большая частота хромосомных аномалий в материале выкидышей объясняет высокую частоту комбинированных аномалий в одном и том же зародыше. Напротив, у новорожденных комбинированные аномалии крайне редки. Обычно в таких случаях наблюдаются комбинации аномалии половой хромосомы и аномалии аутосомы.
В связи с более высокой частотой аутосомных трисомий в материале выкидышей, при комбинированных хромосомных аномалиях у абортусов чаще всего встречаются двойные аутосомные трисомии. Трудно сказать, связаны ли такие трисомии с двойным «нон-дисджанкшн» в одной и той же гамете, или со встречей двух аномальных гамет.
Частота сочетаний различных трисомий в одной и той же зиготе носит случайный характер, что позволяет предположить независимость друг от друга появления двойных трисомий.
Комбинация двух механизмов, приводящих к появлению двойных аномалий, позволяет объяснить появление других аномалий кариотипа, встречающихся при выкидышах. «Нон-дисджанкшн» при образовании одной из гамет в сочетании с механизмами образования полиплоидии объясняет появление зигот с 68 или 70 хромосомами. Сбой первого митотического деления у такой зиготы с трисомией может приводить к таким кариотипам, как 94,XXXX,16+,16+.
Причины
При отсутствии в анамнезе семьи данного заболевания, при абсолютном здоровьем родителей, всё равно есть риск рождения ребенка с данным заболеванием. Науке известно, что клетка человека включает 46 хромосом. В яйцеклетке и сперматозоиде находится по двадцать три хромосомы. Когда они объединяются, то объединяется и количество хромосом. Причины синдрома Эдвардса на сегодняшний день изучены недостаточно.
Исследователи говорят, что вследствие мутаций генов формируется в 18-й паре хромосом одна лишняя. В 2 случаях данной болезни из 100 восемнадцатая хромосома удлиняется, при этом 47-й хромосомы не формируется, а происходит транслокация.
В трех случаях болезни из ста врачи говорят о мозаичной трисомии. Это значит, что лишняя хромосома присутствует только в части клеток организма плода, а не абсолютно во всех. Но по симптомам три описанные варианта болезни сходятся. Только в первом случае течение может быть тяжелым и больше шансов летального исхода.
Показания для тестирования на ломкую Х-хромосому
- Индивиды с задержкой умственного и общего развития, аутизмом
- Индивиды с чертами фрагильной Х-хромосомы
- Индивиды с наличием синдрома фрагильной Х-хромосомы в семейном анамнезе
- Индивиды с наличием в семейном анамнезе недиагностированной задержки умственного развития
- Плоды от матерей-носителей
Геномный импринтинг — процесс, при котором активация гена происходит преимущественно в материнской или преимущественно в родительской хромосоме, но не в обеих хромосомах. Нормальное развитие имеет место лишь в том случае, если присутствуют обе копии (материнская и отцовская) импринтинг-ген. Импринтинг-ген неактивен, значит, активный ген теряет (путем делеции) или получает мутацию, в таком случае плод будет пораженным. Лишь несколько генов могут испытывать импринтинга. Примерами геномного импринтинга может быть синдром Ангельмана и полный пузырный занос (вариант гестационной трофобластической болезни).
Синдром Ангельмана характеризуется тяжелой задержкой умственного развития, атаксической походкой, типичным лицом, пароксизмами смеха и судорогами. Ген синдрома Ангельмана является активным только в материнской унаследованной хромосоме, следовательно, если происходит делеция материнской хромосомы 15 или материнская копия гена имеет мутацию, белковый продукт не образуется и плод будет пораженным.
Синдром Ангельмана также может развиться, если обе копии хромосомы 15 является унаследованными от отца (отсутствие материнской копии хромосомы 15). Это состояние получило название унипарентальной дисомии. Унипарентальная дисомия возникает чаще вследствие потери хромосомы у эмбриона с трисомией или добавления хромосомы у плода с моносомией по этой хромосомой. Каждая из хромосом может быть генетически различной (гетеродисомия) или идентичной (изодисомия), в зависимости от времени возникновения этого феномена — в течение первого или второго мейотического деления, соответственно.
Полный пузырный занос обычно является диплоидным (46, ХХ или Х ¥), но может иметь полностью отцовское происхождение, без материнского хромосомного материала. При таких условиях плод не может развиваться. Полный пузырный занос может сопровождать нормальную многоплодную беременность, но в этом случае возрастает риск материнских осложнений (гипертиреоидизм, преэклампсия, преждевременные роды). В отличие от полного пузырного заноса, частичный пузырный занос обычно является триплоидным (69, ХХХ, 69, ХVV), с дополнительным набором отцовских хромосом.
Триплоидия с дополнительным набором материнских хромосом имеет место при ЗВУР плода, врожденных пороках развития и маленькой плаценте.
Возникновение
У людей с трисомией 18 хромосома 18 или ее часть присутствует трижды (= трисома ) вместо обычно два раза (= дисома ) во всех или некоторых клетках тела. Различают следующие типы:
Бесплатная трисомия 18
У большинства людей с синдромом Эдвардса (около 95%) все клетки тела имеют тройную 18 хромосому. Этот тип синдрома возникает, когда одна из половых клеток содержит дополнительную 18 хромосому. Ошибка возникает, когда пара хромосом 18 не разделяется как обычно (как другие пары хромосом) во время образования яйцеклеток или сперматозоидов. В мейозе такое нерасхождение происходит случайно . Частота свободной трисомии 18 увеличивается с возрастом матери, хотя каждая женщина с детородным потенциалом может ожидать ребенка с трисомией 18 в любом возрасте. Кариотип свободной трисомии 18, 47, XX, + 18 для девушки и 47, XY, + 18 для мальчика.
Мозаичная трисомия 18
В генетике, мозаики являются понимаются наличие нескольких кариотипов в том же самом организме. В мозаичной трисомии 18, которая поражает около 3% людей с синдромом Эдвардса, сосуществуют трисомия и линия дисомных клеток. Некоторые клетки тела имеют нормальные ядра с 2n = 46, другие клетки имеют больные ядра с 47 хромосомами. Таким образом, кариотип мозаики формулируется как 46, XX / 47, XX, + 18 или 46, XY / 47, XY, + 18. После оплодотворения первые деления протекают нормально, так что регулярные митозы приводят к образованию ядер дисомных клеток. Но если разделение сестринских хроматид 18 хромосомы не проводится, ядро клетки получает 18 троекратно. В этом случае нерасхождение происходит в митозе и является источником трисомной клеточной линии. Такое патологическое распределение хромосом одновременно вызывает моносомию 18 в ядре другой (родственной) клетки. Однако формула мозаики игнорирует это событие, поскольку полная моносомия аутосомы фатальна для пораженной клетки.
Чем позже в эмбриогенезе неправильно распределены хроматиды 18 хромосомы, тем меньше возникает трисомных клеток. Чем больше остается дисомных клеток, тем слабее проявляются симптомы синдрома.
Частичная трисомия 18
Этот тип встречается примерно у 2% людей с синдромом Эдвардса. При частичной (частичной) трисомии 18 хромосомы 18, как обычно, присутствуют в двух экземплярах во всех клетках тела, но часть одной из двух хромосом 18 дублируется. В результате одна из 18 хромосом немного длиннее другой, а ядра пораженных клеток трижды содержат генетическую информацию этого участка. У людей с частичной трисомией 18 признаки синдрома не проявляются в высокой степени. Но это ожидание не следует обобщать, а всегда следует рассматривать в каждом конкретном случае, потому что выражение в каждом случае зависит от информационного содержания тройного сегмента хромосомы.
Транслокационная трисомия
Тип, при котором материал трисомной хромосомы из хромосомы 18 прикрепляется к другой хромосоме, очень редок и поэтому почти не упоминается в статистике. Это изменение расположения хромосом называется в генетике транслокацией , форма трисомии 18 называется транслокационной трисомией 18. Кариотип есть, в зависимости от того, какой хромосоме соединение производится, например: 46, XX, т (18; 22) или 46, XY, т (18; 22). При транслокационной трисомии 18 в некоторых случаях «носителем» может быть один родитель . У такого родителя можно обнаружить сбалансированную транслокацию 18-й хромосомы . Кариограмма показывает 45 вместо 46 отдельных хромосом, потому что две хромосомы соединились. Поскольку соответствующий генетический материал не теряется или не добавляется к этой особенности, генетическая информация находится в равновесии (= сбалансированном ), и человек не испытывает трисомии 18. Однако вероятность того, что заинтересованное лицо станет отцом ребенка с транслокационной трисомией 18, увеличивается. Если обе хромосомы 18 соединились друг с другом в результате сбалансированной транслокации у одного из родителей, ребенок, зачатый этим человеком, всегда имеет трисомию транслокации 18/18.
Скрининг на генетические заболевания
Сегодня известно более 11 000 моногенных заболеваний, которые кодируются одним геном (генетически обусловленные) и передаются от родителей их потомкам. Механизм передачи многих генетических болезней объясняется принципами Менделя.
Аутосомно-доминантные моногенные синдромы встречаются с частотой 1: 200 индивидов; заболевание наблюдается у многих поколений, передается потомкам и рецидивирует с частотой 50%. Примерами аутосомно-доминантных моногенных расстройств могут быть:
- ахондроплазия,
- нейрофиброматоз,
- синдром Марфана,
- болезнь Хантингтона,
- семейный полипоз.
Появление аутосомно-доминантных заболеваний у новорожденных от «здоровых» родителей может быть обусловлено следующими причинами:
1. Мозаицизм зародышевых клеток. Мутация может иметь место лишь в популяции зародышевых клеток. Итак, родители являются непораженными, но могут передавать мутацию потомкам.
2. Новые мутации. Рост возраста родителей ассоциируется с увеличением риска аутосомно-доминантных расстройств (ахондроплазии, танатофорной дисплазии, нейрофиброматоза, синдрома Аперта — краниосиностоз). Риск рецидивов у других детей не увеличивается.
3. Вариабельна экспрессия. Тяжесть заболевания может варьировать, и родители могут не распознать мягкие и субклинические мутации.
4. Уменьшенная пенетрантность. Родители могут иметь аномальный ген без клинических проявлений заболевания.
5. Неверное отцовство. Частота неверного отцовства достигает 15%.
Аутосомно-рецессивные моногенные заболевания проявляются в многочисленных родственников при наличии двух пораженных аллелей. Если оба родителя являются носителями пораженного гена, риск заболевания у потомства равен 25% при каждой беременности. Аутосомно-рецессивные заболевания включают кистозный фиброз, серповидно-клеточную анемию, фенилкетонурию, болезнь Тея-Сакса, Канавана и др.
При Х-сцепленных рецессивных синдромах (гемофилия и др.) мать-носитель пораженного гена передает его своим сыновьям. Итак, 50% сыновей могут быть больными и 50% дочерей будут носителями этого гена. Редкие Х-доминантные синдромы могут передаваться от каждого родителя каждому ребенку подобно аутосомно-доминантных синдромов. Фенотип может сильно варьировать, что связано со смешанной пенетрантностью, лионизацией (гетерохроматизацией) Х-хромосомы (синдром ломкой Х-хромосомы) и геномным импринтингом.
Экспансия тринуклеотидных повторов. Некоторые гены содержат участки тройных повторов (например, ССС). Такие участки являются нестабильными и могут увеличиваться в следующих генерациях, этот феномен получил название антиципации. Количество повторений определяет степень поражения индивида. Экспансия тринуклеотидных повторов составляет основу многочисленных генетических расстройств, таких как синдром ломкой (фрагильной) Х-хромосомы, миотоническая дистрофия и болезнь Хантингтона.
Синдром ломкой (фрагильной) Х-хромосомы является наиболее частой причиной семейной задержки умственного развития. Пораженные мужчины имеют типичные черты: большие уши, выступающая челюсть, большие яички, аутичное поведение, легкая или умеренная умственная отсталость. Женщины обычно менее поражены в связи с инактивацией Х-хромосомы.
Ген ломкой Х-хромосомы локализуется в Х-хромосоме и имеет три нуклеотидные повтора (ССС). Нормальные индивиды имеют 6-50 повторов, непораженные носители женского пола могут иметь 50-200 повторов, которые могут распространяться на мейоза до полной мутации при наличии более 200 повторов. Если имеет место полная мутация, ген инактивируется путем метилирования; плод будет пораженным. Тяжесть заболевания зависит от степени Х-инактивации у женщин, степени метилирования и мозаицизма размера повторов.
Женщины-носители премутации имеют 50%-й риск передачи гена с экспансией. Мужчины с премутациею фенотипически являются нормальными, но все их дочери будут носителями премутации. В случае трансмиссии мужчинам количество повторов остается стабильным. Тест на ломку Х-хромосому выполняется с целью выявления количества повторов и степени метилирования.
Возможные осложнения при родах
Во время беременности ребенком с трисомией 18 наблюдается избыток околоплодных вод и единственная пупочная артерия, которая мешает достаточному поступлению кислорода, следствием чего является состояние асфиксии у новорожденных.
Также часто встречается низкая плацентация, что ведёт к неправильному предлежанию и мешает правильному опущению головы ребенка.
Эта патология затрудняет естественные роды и иногда делает их невозможными, приходится выполнять срочное кесарево сечение. При проведении операции эта аномалия приводит к обильной кровопотери и может быть опасна для жизни матери.
Дети с трисомией 18 рождаются недоношенными или переношенными, масса тела обычно составляет 2100-2300 грамм.
Клинические признаки трисомии 18
Внешние аномалии:
- долихоцефалия-кости черепа узкие и длинные;
- микроцефалия-диспропорция размера головы в соотношении с размерами туловища;
- неправильное развитие ушной раковины. Уши находятся ниже уровня глаз, отсутствуют выпуклости хряща, который формирует ушную раковину;
- расщепление неба. Происходит вследствие незаращения двух частей верхней челюсти во время внутриутробного развития.
- искажения лица;
- сколиоз;
- косоглазие;
- деформации суставов;
- стопа-качалка, или дисплазия стоп.
Дисфункции внутренних органов:
- пороки сердца;
- недостаточный тонус мышц;
- отсутствие сосательного рефлекса;
- недоразвитость почек; пупочные и паховые грыжи;
- атрофия мозговых извилин; умственная отсталость, задержки в развитии.
Методы диагностики
Инвазивные:
- биопсия тканей плода плацентоцентез — забор плацентарных клеток, который проводится во втором триместре беременности;
- амниоцентез — забор околоплодных вод для исследования содержания гормонов, аминокислот и ферментов, которые отвечают за развитие и рост плода, проводится на 16 неделе беременности.
- кордоцентез — забор крови из пуповины.
Проводится с целью выявления хромосомных и гормональных аномалий, исследование выполняется на 18-24 неделях беременности; биопсия хориона-забор клеток зародышевой оболочки. Проводится на 10-11 неделе беременности.
Неинвазивные:
- УЗИ (трансабдоминальное и трансвагинальное исследования). Трансвагинальное УЗИ лучше всего проводить на самых ранних сроках, а трансабдоминальное-во 2 и 3 триместрах.
- анализ родословной обоих родителей.
- диагностика сывороточных маркеров плода (анализ крови, который позволяет узнать вероятность развития нарушения нервной трубки у ребенка и выявить хромосомные аномалии)
Применяется, чтобы сделать анализ внеклеточной ДНК развивающегося плода по крови матери. Делать рекомендуется с 9 недели беременности, когда в кровь женщины уже проникло достаточное количество клеток крови плода.
Лаборатория Медикал Геномикс рекомендует расширенный неинвазивный пренатальный тест VERAGENE
Прогноз
Около 95% таких беременностей не заканчиваются рождением живого ребенка. Основные причины смерти включают апноэ и сердечные аномалии. Предсказать точный прогноз при беременности или в периоде новорожденности невозможно . Половина живых младенцев не доживает до первой недели жизни. Средний срок службы составляет от пяти до 15 дней. Около 8–12% младенцев живут дольше 1 года. Один процент детей доживает до 10 лет. Однако эти оценки могут быть пессимистичными; ретроспективное канадское исследование 254 детей с трисомией 18 продемонстрировало 10-летнюю выживаемость 9,8%, а другое обнаружило, что 68,6% детей с хирургическим вмешательством выжили в младенчестве.
Причины и признаки
Синдром трисомия 18, как именуют еще патологию,представляет собой совокупность патологических состояний с неблагоприятным прогнозом для жизни. Всему виной появление 18 хромосомы. Кариотип больной девочки 47,XX, 18+ и тип мутации больного мальчика 47,XY, 18+.
Причины развития патологии:
- фактором возникновения служит образование лишней хромосомы в кариотипе при синдроме эдварса. Таким образом, вместо двух полноценных хромосом формируются три;
- 18 хромосома присутствует в организме родителя еще до зачатия;
- также наблюдается мозаицизм в некоторых случаях. Это означает, что не расхождение проявилось на раннем этапе развития зародыша.
Что конкретно стимулирует данное явление, ученым на данный момент неизвестно. Однако есть общие факторы риска, которые провоцируют возникновение генетических аномалий:
- возраст женщины старше 45;
- генетическая склонность;
- употребление наркотических веществ, злоупотребление спиртным и курение;
- продолжительный прием мощных медикаментозных препаратов;
- заболевания, передающиеся половым путем;
- радиация.
Перечисленные выше факторы являются только предполагаемыми, но не доказанными научно. Многие интересуются, какова частота встречаемости данной аномалии. На семь тысяч малышей приходится один ребенок с таким диагнозом. В связи с тем, что прогноз и качество жизни таких пациентов крайне негативные, целесообразно выявить патологию еще до оплодотворения.
Любопытно, что девочки с таким синдромом появляются на свет в три раза чаще, чем мальчики.
Что после рождения?
Младенцы, рожденные с синдромом Эдвардса, обычно рождаются недоношенными, с очень низкой массой тела при рождении и требуют интенсивной терапии с самого рождения. Дефекты, связанные с возникновением этого заболевания, могут включать многие системы и органы, в том числе: пороки сердца, гортань, диафрагмальную грыжу, стриктуру пищевода, гидронефроз и грыжу позвоночника.
Выживаемость новорожденных с синдромом Эдвардса составляет около 50% в первые недели жизни. Однако до 90% детей умирают в возрасте до 1 года.
Обычно кормление ребенка необходимо через зонд, введенный в желудок или энтерально, из-за нарушения сосательного и глотательного рефлексов. Малыши также находятся под наблюдением кардиолога, невролога, нефролога, генетика, а также нуждаются в реабилитации для улучшения работы органов движения.
Симптомы
Семейной паре нужно понимать, что такое болезнь эдвардса, и какие страдания она способна принести родителям и ребенку. Симптоматика крайне не радостна. Первые признаки будут давать о себе знать уже после рождения:
- Асфиксия.
- Волчья пасть.
- Опущение век (птоз).
- Слабоумие.
- Нарушение глотания.
- Нарушение дыхания.
- Сердечные и сосудистые пороки.
- Заболевания системы пищеварения.
- Нарушения работы мочеполовой системы.
- Проблемы с психикой.
Учитывая все перечисленные симптомы, можно подвести итог, что больной ребенок никогда не станет полноценным членом общества, как бы этого не хотелось. Жизненный путь у него будет коротким, наполненным постоянными преградами. При этом больной не будет понимать и осознавать своего тяжелого состояния из-за тяжелой степени олигофрении.
Но это далеко не все проявления. Эдвардс описал свыше ста тридцати тяжелых симптомов данного недуга.
Лечение
Почти всегда приобретенные при формировании организма пороки являются несовместимыми с жизнью, поэтому лечение сводится к тому, чтобы облегчить симптоматику, продлить жизнь и максимально нормализовать физиологические функции.
Дети, страдающие синдромом Эдвардса, довольно часто болеют синуситом, конъюнктивитом, средним отитом и инфекционными процессами в мочеполовой системе. В связи с этим основным врачом, который обеспечивает поддержание их жизни, является педиатр. Он регулярно проводит осмотры, дает рекомендации по правильному уходу, следит за достаточным питанием.
Хирургическое исправление отклонений слишком опасно и неоправданно, поэтому проводится крайне редко.